Русский Медицинский Сервер / Лечение в Италии / Центр лечения редких заболеваний в Милане / Синдром Барде-Бидля - лечение в Италии
Синдром Барде-Бидля - лечение в Италии
Синдром Барде-Бидля (альтернативное название - синдром Лоренса-Муна) - генетически гетерогенное заболевание, различают 19 типов BBS. К первичным симптомам BBS относятся деградация сетчатки глаза, ожирение, умственная отсталость, полидактилия, аномалии развития почек, гипогонадизм. Для постановки диагноза необходимо наличие четырех первичных симптомов. Ко вторичным симптомам относятся диабет, фиброз печени, гипертензия, атаксия, аносомия, потеря слуха, также описано несколько случаев BBS, ассоциированных с болезнью Гиршпрунга
Синдром Барде-Бидля принято считать аутосомно-рецессивным заболеванием, однако было показано, что для клинической манифестации некоторых форм BBS необходимы рецессивные мутации в одном из шести локусов плюс дополнительная мутация во втором локусе, такой тип наследования называют трехаллельным или рецессивным наследованием с модификатором пенетрантности.
Популяционная частота синдрома Барде-Бидля в Европе среди новорожденных невысокая 1: 160 000. Наиболее часто встречаются мутации в генах BBS1 и BBS10 (32,6%), а также в BBS12 (10,4%).
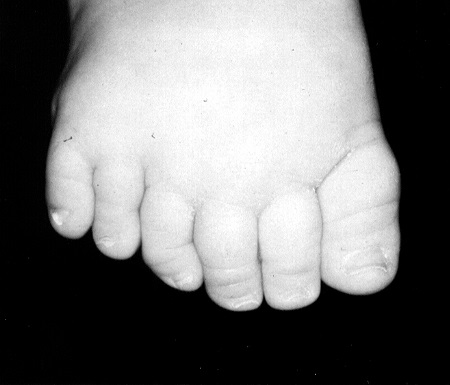
Заболевание имеет наследственную природу с аутосомно-рецессивным типом наследования. Характеризуется большой вариабельностью проявлений. Чаще наблюдается 3 или 4 признака (неполная форма), реже — пять (полная форма). Из основных симптомов наиболее часто встречаются пигментный ретинит и другие изменения сетчатки (93%), ожирение (90%) и умственная отсталость (87%); полидактилия обнаруживается у 60—70% больных.
BBS1 вызван мутациями в гене, расположенном на хромосоме 11q13.2, содержащем 17 экзонов, данный ген кодирует один из семи белков, которые образуют ядро стабильного протеинового комплекса, необходимого для цилиогенеза.
BBS10 вызван мутациями в гене C12ORF58, расположенном на хромосоме 12q21.2, состоящем из 2 экзонов. Ген C12ORF58 кодирует белок BBS10, который принадлежит к подсемейству шаперонов и участвует в процессе локализации первичных цилий на поверхности жировых клеток во время дифференцировки преадипоцитов.
Часто наблюдаются дефекты развития почек. Спектр поражения почек широко варьирует: гипоплазия, кортикальные и медуллярные кисты, различные формы дисплазий, перигломерулярный и интерстициальный фиброз, пролиферация мезангия. При гистологическом исследовании яичек обнаруживаются перитубулярный фиброз, связанный с утолщением базальной мембраны, уменьшение числа сперматогониев, гиалиноз канальцев. Сравнительно редко встречаются пороки сердца.
Пигментная дегенерация сетчатки проявляется к 6—7 годам снижением зрения и с возрастом неуклонно прогрессирует. Возможна и другая патология зрения — макулярная дегенерация, катаракта, миопия, атрофия зрительных нервов, микрофтальмия, отсутствие всей радужки или большей ее части (аниридия). К 30 годам большинство больных практически полностью слепнут.
Ожирение проявляется на 1—2-м году жизни, быстро прогрессирует, достигая III—IV степени.
Изменения кистей и стоп характеризуются увеличением числа пальцев (полидактилия), возможно сочетание со сращением соседних пальцев (синдактилия) и их укорочением (брахидактилия).
У большинства больных отмечается умственная отсталость от легкой дебильности до идиотии. У детей с полной формой заболевания чаще имеется глубокая умственная отсталость (имбецильность или идиотия), а при неполных формах — лишь легкие нарушения познавательных функций. В то же время при этом синдроме не исключен и нормальный интеллект. Признаки органического поражения ц.н.с. проявляются инертностью психических процессов, выраженной астенией в сочетании с головными болями, головокружениями, некоторым ухудшением памяти и др. Иногда возникают судороги, возможны экстрапирамидные расстройства.
Диагноз при полной форме синдрома не представляет затруднений. При неполных формах синдрома может возникнуть необходимость в разграничении его с синдромом Альстрема, для которого также характерны пигментная дегенерация сетчатки, ожирение, поражение почек, но, кроме того, имеется сахарный диабет, что позволяет дифференцировать эти два состояния.
Необходима также дифференциальная диагностика с адипозогенитальной дистрофией, для которой характерны ожирение и гипогенитализм, но отсутствуют полидактилия, олигофрения и пигментная дегенерация сетчатки.
Специфического лечения нет. Ожирение корригируют назначением диеты. Полидактилию устраняют оперативным путем. Разработаны методы предупреждения прогрессирования снижения зрения вследствие пигментной дегенерации сетчатки.
Вероятность повторного рождения больного ребенка в семье составляет 25%, что относится к категории высокого генетического риска. При планировании деторождения в семье, где уже есть больной ребенок, необходимо медико-генетическое консультирование.
! Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку - так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
+7 (925) 50 254 50 – срочное лечение в Италии