Русский Медицинский Сервер / Лечение в Италии / Центр лечения редких заболеваний в Милане / Синдром Фрейзера - лечение в Италии
Синдром Фрейзера - лечение в Италии
Синдром Фрейзера является редкой формой генетически детерминированных пороков развития с аутосомно-рецессивным типом наследования. Частота унаследования колеблется в диапазоне 15-25%. Выживаемость при данном типе патологии низкая, дети умирают в возрасте до одного года.
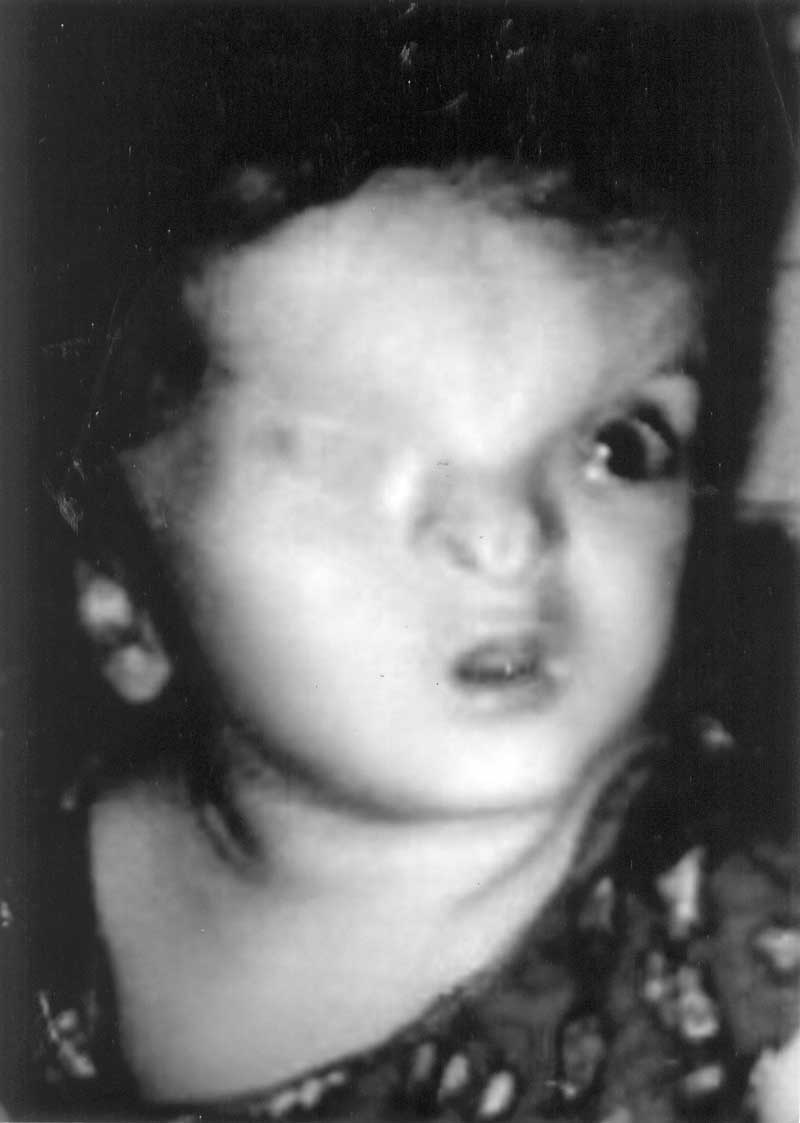
Синдром Фрейзера характеризуется множественными пороками развития, в том числе криптофтальмией, синдактилией рук и ног, половыми аномалиями, и часто ассоциирован с пороками развития почек, уха, носа, гортани, скелета. Одним из главных диагностических признаков синдрома Фрейзера является криптофтальмия, но она не является обязательной. В 1962 году Джордж Фрейзер был первым, кто диагностировал у двух братьев криптофтальмию, связанную с другими отклонениями в развитии.
Со стороны лор органов лор пациентов беспокоит снижение слуха, за счёт врождённой аномалии развития наружного уха (ушной раковины), сужения (атрезии) наружного слухового прохода, недоразвития барабанной полости среднего уха и ячеистой структуры клеток сосцевидного отростка (склеротический тип строения сосцевидного отростка).
Со стороны глаза отмечается выраженное сужение глазной щели (криптофтальм) и недоразвитие (сужение) или полное отсутствие (атрезия) слёзных протоков. Со стороны верхних дыхательных путей отмечается сужение просвета гортани, что провоцирует нехватку воздуха или удушье.
При осмотре глотки отмечается «готическое небо», может сопровождаться расщелиной губы и твёрдого нёба. Встречаются пациенты со сросшимися пальцами (синдактилия). Со стороны желудочно-кишечного тракта отмечается аномалия развития брыжейки тонкой кишки, провоцирующая спаечную болезнь.
Со стороны мочевыделительной системы отмечается гипоплазия (недоразвитие) почек, что сопровождается нарушением их фильтрационной способности. Со стороны генитальной сферы у женщин возможно сращение малых половых губ, увеличение в размере клитора. Матка чаще двурогая. Встречаются так же аномалии развития и проходимости яичников.
Ген SRY. После описания синдрома Клайнфелтера (мужской фенотип с кариотипом 47, XXY) и синдрома Шерешевского-Тернера (женский фенотип с кариотипом 45, X) стало ясно, что мужская дифференцировка эмбрионов млекопитающих зависит от наличия Y-хромосомы, а не от количества Х-хромосом. Ген Y-хромосомы, ответственный за половую дифференцировку по мужскому типу, был назван TDF (тестикулодетерминирующий фактор), однако идентифицировать его оказалось не так просто.
Из-за того, что Х- и Y-хромосомы спариваются и расщепляются в процессе мейоза подобно аутосомам, было сделано предположение, что у Х- и Y-хромосом существуют аутосомоподобные регионы в дистальных отделах плеч, или псевдоаутосомные регионы, обеспечивающие спаривание и расщепление во время мейоза. Magenis и соавт. в 1982 г. описали мужчин с кариотипом 46, XX, у которых имелась транслокация региона Yp в один из регионов Хр, продемонстрировав таким образом один из механизмов первичного несоответствия пола.
Наличие конъюгации и кроссинговера между псевдоаутосомными регионами Yp ближе к центромере приводит к переносу TDF на Х-хромосому. Этот феномен способствовал созданию делеционных карт Y-хромосомы и выявлению гена SRY, который и оказался тем самым фактором TDF. Открытие мутаций гена SRY при чистой дисгенезии гонад с кариотипом XY подтвердило роль SRY в качестве гена-кандидата на TDF.
Синдром Фрейзера характеризуется полосковидными гонадами, аномалиями развития почек и нефротическим синдромом. Его причина — мутации гена WT1. Этот ген кодирует ДНК-связывающий фактор транскрипции, имеющий 24 изоформы за счет альтернативного сплайсинга. Донорные сайты альтернативного сплайсинга на конце 9-го экзона обеспечивают инсерцию (+) или делецию (-) трех аминокислот — лизина, треонина и серина (так называемая KTS-последовательность) — между третьим и четвертым «цинковыми пальцами» фактора транскрипции. Больные с кариотипом XY, страдающие синдромом Фрейзера, характеризуются утратой +КТS-изоформы WT1, приводящей к снижению образования белка SRY в урогенитальной складке и несоответствию пола генотипу XY. Фенотип у таких больных женский, с дисгенезией половых желез (полосковидные гонады), структур, подобных мюллеровым протокам, и нефропатией.
! Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку - так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
+7 (925) 50 254 50 – срочное лечение в Италии