Русский Медицинский Сервер / Лечение в Италии / Центр лечения редких заболеваний в Милане / Синдром Донохью (лепречаунизм) - лечение в Италии
Синдром Донохью (лепречаунизм) - лечение в Италии
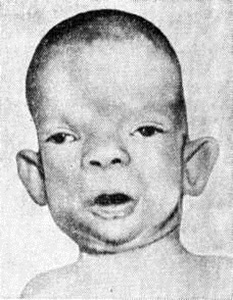
Синдром Донохью, он же лепреконизм, синонимы – лепречаунизм, от ирландского leprechaun – гном, дефект рецептора инсулина) – наследственное заболевание: гротескные черты лица, гипертелоризм (широко расставленные глаза), экзофтальм (смещение глазного яблока вперед за пределы глазницы), толстые губы, крупные низко расположенные уши, широкий кончик носа, избыточное оволосение, увеличение молочных желез, клитора, половых губ у девочек и полового члена у мальчиков.
Заболевание впервые описано в 1948 год американский учёным Донохью (W. L. Donohue). Лепречаунизм наследуется по аутосомно-рецессивному типу, сцепленному с полом; возможны семейные случаи заболевания. Болеют девочки.
Морфологически обнаруживаются кисты в яичниках, гиперплазия панкреатических островков, гипертрофия и кальциноз почек, накопление гликогена и железа в печени, гиперплазия ткани молочной железы.
Описаны грыжи разной локализации, задержка костного возраста. Психомоторное развитие задержано. При обследовании выявляют гиперинсулинемию, гипогликемию, аминоацидурию; в клетках печени накапливаются гликоген и железо; на аутопсии – гиперплазия и фолликулярные кисты яичников, гиперплазия островкового аппарата поджелудочной железы, гипертрофия и кальциноз почек. Наследуется аутосомно-рецессивно.
Отмечается гиперкальциемия (смотри полный свод знаний), повышенная чувствительность к инсулину, несколько повышенное выделение 17-кетостероидов (смотри полный свод знаний) с мочой.
Синдром характерен для детей, родившихся от близкородственных браков, наследование аутосомно-доминантное. Основные признаки:
- внутриутробная постнатальная задержка развития
- низкорослость
- гротескное лицо
- липодистрофия
- мышечная слабость
- acanthosis nigricans
- гиперандрогения яичникового происхождения
- преждевременное половое развитие
- гипертрофическая кардиомиопатия
- смерть в молодом возрасте
Этиология ключает в себя мутацию гена рецептора инсулина, возможно, в сочетании с дефектами рецепторов ИФР-I и эпидермального фактора роста.
Диагноз основывается на данных анамнеза и клинические, картины болезни. Лаб. исследования имеют вспомогательное значение. Дифференциальный диагноз проводится с различными эмбриопатиями и фетопатиями, врождённой гипотрофией, синдромом мальабсорбции.
Прогноз неблагоприятный, большинство детей умирает в раннем возрасте.
! Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку - так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
+7 (925) 50 254 50 – срочное лечение в Италии