Русский Медицинский Сервер / Лечение в Италии / Центр лечения редких заболеваний в Милане / Лейкодистрофия - лечение в Италии
Лейкодистрофия - лечение в Италии
Лейкодистрофии (прогрессирующие склерозы мозга) - наследственно-дегенеративные заболевания нервной системы, обусловленные генетическим дефектом энзимов, участвующих в метаболизме липидов, преимущественно миелина, и характеризующиеся прогрессирующим распадом миелина и вторичной гибелью нервных клеток. Название болезни дано Bielshowsky и Henneberg в 1928 г. при описании семейных форм прогрессирующих диффузных склерозов мозга. В мировой литературе к 1960 г. описано немногим более 120 случаев лейкодистрофий, половина из которых имела семейный характер. Основным типом наследования лейкодистрофий является аутосомно-рецессивный, реже может иметь место рецессивный, сцепленный с полом тип.
Лейкодистрофия получила свое название в связи с поражением белого вещества мозга (с греческого leukos — белый). Различают около 60 разновидностей лейкодистрофии, определяющихся видом генной аномалии и возрастом манифестации клинических проявлений. Наряду с отдельными воспалительными поражениями ЦНС (например, лейкоэнцефалитом Шильдера) лейкодистрофия относится к синдрому диффузного склероза мозга. При этом доминирующее поражение миелина сближает ее с демиелинизирующими заболеваниями (рассеянным склерозом, РЭМ и пр.), а отдельные формы можно отнести к липидозам.
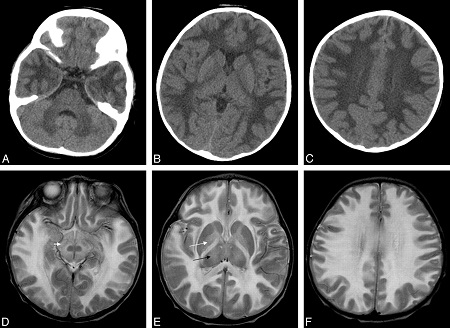
Головной мозг поражается диффузно, симметрично страдают оба полушария, ствол головного мозга, мозжечок. Значительные изменения постоянно обнаруживаются в пирамидных путях. Нередко в процесс вовлекается спинной мозг. Гистологически лейкодистрофии характеризуются распадом миелина и в ряде случаев накоплением образующихся при распаде веществ в паренхиматозной ткани, глии или макрофагах.
Серое вещество изменяется в меньшей степени. В мозговой ткани выявляется наличие глобоидных клеток, спонгиозное состояние промежуточных участков между белым и серым веществом головного мозга и некоторые другие изменения, присущие отдельным формам лейкодистрофий. В основе генеза лейкодистрофий лежит генетически обусловленный дефект энзимов, ответственных за обмен липидов, входящих в состав миелина, что приводит к его ускоренному преждевременному распаду ("дисмиелинизация").
Продукты нарушенного обмена липидов могут накапливаться не только в мозге, но и в других органах, крови и ликворе. При классификации лейкодистрофий учитываются гистологические особенности отдельных форм, биохимические и клинические признаки лейкодистрофий. Основными формами лейкодистрофий являются: метахроматическая лейкодистрофия Шольца - Гринфильда, глобоидно-клеточная лейкодистрофия Краббе, лейкодистрофия Галлевордена - Шпатца, суданофильная лейкодистрофия Пелицеуса - Мерцбахера, спонгиозная дегенерация белого вещества мозга - болезнь Канавана - ван Богарта - Бертрана; лейкодистрофия с наличием диффузной волокнистой формации Розенталя - болезнь Александера.
В большинстве случаев лейкодистрофия дебютирует в раннем детском возрасте. Новорожденные, как правило, выглядят здоровыми. Определенный период они нормально развиваются, а затем постепенно возникают различные неврологические симптомы, отличающиеся неуклонным прогрессированием. Скорость нарастания симптомов тем выше, чем раньше манифестировала лейкодистрофия. Ведущими проявлениями выступают прогрессирующая олигофрения, ухудшение зрения, тугоухость, эписиндром, спастические парезы.
Первыми симптомами лейкодистрофии могут быть атаксия, мышечно-тонические расстройства (гипо- или гипертонус, мышечные подергивания), экстрапирамидные проявления, изменения поведения. Затем возникают эпиприступы, бульбарные проявления, снижается слух и зрение, отмечается интеллектуальное снижение с постепенной утратой ранее приобретенных навыков. Сенсорные расстройства не характерны. На поздних этапах развития болезни наблюдаются параличи, выраженная олигофрения, грубое расстройство глотания, амавроз, глухота. В терминальной фазе обычно отмечается децеребрационная ригидность.
Виды лейкодистрофии:
- Метахроматическая лейкодистрофия в зависимости от манифестации имеет 4 варианта. Врожденный вариант дебютирует в первые 1-3 мес. жизни задержкой развития и судорожным синдромом; дети не достигают возраста 1 года. Позднедетский вариант метахроматической лейкодистрофии начинается в период от 1 до 3 лет с мышечной гипотонии и слабости, атаксии, задержки психического развития (ЗПР). Затем формируется спастическая тетраплегия, афазия, псевдобульбарный синдром. В редких случаях пациенты доживают до 10-летнего возраста. Ювенильный вариант манифестирует в 4-6 лет и длится в среднем 7 лет. Взрослый вариант дебютирует в третьей декаде жизни, иногда позднее, продолжительность жизни пациентов от начала клиники варьирует в пределах 10-20 лет.
- Суданофильная лейкодистрофия наследуется сцеплено с Х-хромосомой и имеет несколько разновидностей. Лейкодистрофия Пелицеуса-Мерцбахера может стартовать на 1-ом году жизни или в 3-4 года. Первым признаком является крупноразмашистый нистагм, позже возникает ЗПР, мозжечковая атаксия, гиперкинезы, парезы. Наибольшее прогрессирование происходит в возрасте до 10 лет, затем заболевание принимает замедленное течение с длительными ремиссиями. Пациенты могут жить до зрелого возраста. Адренолейкодистрофия — вариант, при котором лейкодистрофия сочетается с надпочечниковой недостаточностью. Характеризуется прогрессирующим течением с летальным исходом спустя 6-8 лет от начала клиники.
- Глобоидно-клеточная лейкодистрофия (болезнь Краббе) — липоидоз с накоплением в очагах демиелинизации галактоцереброзида и образованием больших округлых глобоидных клеток. Раннедетский вариант развивается в первом полугодии жизни с гипервозбудимости и периодической гипертермии, задерживается психомоторное развитие, нарастает тонус мышц, затем развивается спастический тетрапарез, олигофрения, эписиндром, возможен опистотонус. В годовалом возрасте наступает летальный исход. Позднедетский вариант более редкий, манифестирует ухудшением зрения.
- Спонгиозная дегенерация Ван-Богарта-Бертрана характеризуется эписиндромом, гиперсомнией, выраженной гидроцефалией с увеличением размеров головы, вызывающей амавроз атрофией зрительных нервов. Резкая внутричерепная гипертензия приводит к расхождению черепных швов, регистрируемому при рентгенографии черепа. Пациенты с этой формой лейкодистрофии погибают до 3-летнего возраста.
- Болезнь Александера (лейкодистрофия с волокнистой формацией) обусловлена мутацией гена, ответственного за синтез GFAP белка. В результате происходит накопление в клетках глии аномального GFAP белка, содержащего волокна Розенталя. Неонатальный вариант имеет тяжелое течение с летальным исходом к концу 1-го года. Инфантильный вариант встречается примерно в половине случаев, проявляется в первые 1-2 года жизни ЗПР, затем присоединяются спастические парезы, атаксия, гидроцефалия. Дети погибают спустя несколько лет. Ювенильная лейкодистрофия Александера дебютирует в период от 4-х до 10-летнего возраста, протекает с преимущественно стволовой симптоматикой. Продолжительность жизни колеблется в пределах 10-30 лет. Взрослый вариант отличается поздней манифестацией и относительно медленным течением в пределах 10 и более лет.
- Лейкодистрофия Галлервордена-Шпатца чаще всего стартует в 10-летнем возрасте. Проявляется дисфункцией стриопаллидарной системы, затем на фоне гиперкинезов прогрессирует тетрапарез, развивается гемералопия и пигментный ретинит, наблюдается снижение интеллекта, возникают эпиприступы.
Фактически единственным методом лечения лейкодистрофий в настоящее время является аллогенная трансплантация костного мозга (или пуповинной крови) от здорового донора. В случае успеха она может привести к нормализации уровня недостающего белка, а значит, к увеличению продолжительности и качества жизни. Так, известны случаи использования трансплантаций для лечения адренолейкодистрофии, метахроматической лейкодистрофии и глобоидно-клеточной лейкодистрофии.
В то же время использование трансплантаций при лейкодистрофиях связано с серьезными ограничениями. Очень важно провести трансплантацию как можно раньше, до развития заметных неврологических нарушений. Действительно, трансплантация не позволяет «исправить» уже возникшие поражения центральной нервной системы, а только останавливает или замедляет их дальнейшее прогрессирование. Но при этом необходимо учитывать также скорость развития неврологических поражений.
Так, при наиболее быстроразвивающихся формах лейкодистрофий часто не удается избежать гибели или тяжелой инвалидизации больного даже после трансплантации. Это связано с тем, что после трансплантации проходит еще некоторое время (так, при некоторых лейкодистрофиях речь может идти о 12 или даже 24 месяцах) до того момента, как работа донорских клеток приведет к нормальному функционированию миелина. И все это время развитие болезни будет продолжаться. Поэтому при формах с очень ранним началом болезни надежды связаны в основном с теми трансплантациями, которые проведены до появления клинических симптомов (например, если у старшего ребенка в семье уже была обнаружена лейкодистрофия и поэтому младшему ребенку произведена ранняя диагностика). При более медленном развитии болезни шансы на успех повышаются.
! Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку - так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
+7 (925) 50 254 50 – срочное лечение в Италии