Русский Медицинский Сервер / Лечение в Италии / Центр лечения редких заболеваний в Милане / Болезнь Ли - лечение в Италии
Болезнь Ли - лечение в Италии
Ли болезнь (D. Leigh, современный англ. невролог; синоним инфантильная форма болезни Вернике) — редкое прогрессирующее заболевание центральной нервной системы, проявляющееся в раннем детском возрасте и характеризующееся симптомами поражения серого вещества головного и спинного мозга.
В основе болезни лежит генетически детерминированное нарушение обмена тиамина, наследуемое чаще по аутосомно-рецессивному типу. Вследствие нарушения превращения тиаминпирофосфата в тиаминтрифосфат снижается содержание тиамина во многих отделах ц.н.с. — в стволе мозга, базальных ганглиях, мозжечке, спинном мозге. В печени определяется дефицит пируваткарбоксилазы, что приводит к накоплению пирувата и лактата и нарушениям в цикле трикарбоновых кислот.
Известно по крайней мере четыре генетически детерминированных нарушения, лежащие в основе болезни Ли: дефицит пируватде-гидрогеназного комплекса, комплекса I, комплекса IV (СОХ) и комплекса V (АТФаза). Эти нарушения могут возникать спорадически или наследуются по аутосомно-рецессивному типу, как в случае дефицита комплекса IV (СОХ) по Х-сцепленному типу (дефицит пируватдегидрогеназы Е1), или по материнскому типу наследования, как в случае дефицита комплекса V (АТФаза 6, мутация в нуклеотиде 8993). В большинстве случаев заболевание проявляется в младенческом возрасте с трудностей при приеме пищи, нарушения глотания, рвоты и дефицита массы тела.
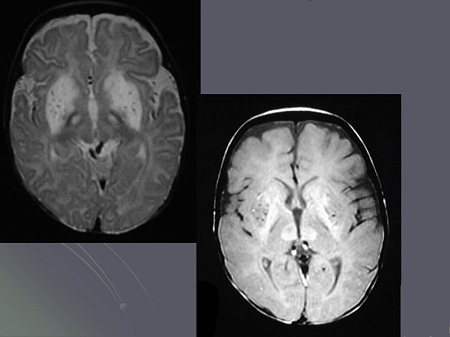
Болезнь Ли характеризуется демиелинизацией , глиозом , некрозом , уменьшением количества нейронов и пролиферацией капилляров в определенных отделах головного мозга. В порядке убывания тяжести поражения эти отделы располагаются следующим образом: базальные ганглии , ствол мозга , мозжечок , кора головного мозга . В основе болезни может лежать недостаточность нескольких ферментативных комплексов. Чаще всего обнаруживается недостаточность цитохром-C-оксидазы, затем недостаточность НАДФ-коэнзим Q-редуктазы , недостаточность пируватдегидрогеназы и недостаточность пируваткарбоксилазы . Из генетических дефектов наиболее часто находят мутации ядерного гена SURF1 , который кодирует фактор биогенеза цитохром-С-оксидазы , и мутации гена митохондриальной ДНК, кодирующего АТФазу.
Морфологические изменения в нервной системе характеризуются поражением серого вещества ствола мозга, базальных ганглиев, четверохолмия, таламуса, мозжечка, спинного мозга. Патология нейронов варьирует от острого клеточного отека до полного хроматолиза. Выраженная пролиферация астроцитов, микроглии, эндотелия сосудов обнаруживается не только в сером, но и в белом веществе.
Возможны задержка двигательного и речевого развития, генерализованные приступы, слабость, гипотония, атаксия, тремор, признаки поражения пирамидных путей и нистагм. Характерно периодическое дыхание в сочетании с вздохами и всхлипываниями, позволяющее предположить дисфункцию ствола. У некоторых пациентов отмечаются наружная офтальмоплегия, птоз, атрофия зрительных нервов и снижение остроты зрения. На КТ или МРТ могут быть билатеральные симметричные области пониженной плотности в базальных ганглиях. Морфологические изменения включают фокальные симметричные области некроза в таламусе, базальных ганглиях, сером веществе покрышки среднего мозга, перивентрикулярных и периакведуктальных областях ствола и в задних канатиках спинного мозга.
Возраст начала заболевания варьирует от 1 нед. до 2 лет. К типичным начальным проявлениям относятся задержка темпа психомоторного развития, дыхательные нарушения по типу диспноэ, тахипноэ, апноэ, дыхания Чейна — Стокса. Дети становятся вялыми, сонливыми, снижается мышечный тонус, расстраивается координация движений, появляется тремор конечностей, усиливающийся при возбуждении и целенаправленных движениях.
По мере течения заболевания гипотония мышц может сменяться их спастичиостью, утрачиваются двигательные навыки, появляются миоклонические подергивания мышц. Сухожильные рефлексы снижены или повышены. Характерны различные зрительные и глазодвигательные расстройства: нарушение цветовосприятия, снижение зрения вплоть до слепоты, атрофия зрительного нерва, нистагм, офтальмоплегия, дискоординированные движения глазных яблок, косоглазие, нарушение реакции зрачков на свет, миоз, мидриаз. Могут наблюдаться судороги, глухота, парезы конечностей вследствие поражения периферических нервов. Наряду с этим отмечаются расстройства функции печени, сопровождающиеся снижением аппетита, рвотой, потерей массы тела. У некоторых больных развивается кардиомиопатия.
Диагноз в амбулаторных условиях затруднен. Предположительный диагноз ставят на основании прогрессирующего нарастания неврологических расстройств, указывающих на избирательное поражение серого вещества головного и спинного мозга и наличия повторных случаев заболевания в семье. Для уточнения диагноза больного необходимо госпитализировать. Примерно у 30% больных в цереброспинальной жидкости обнаруживают увеличение содержания белка и невысокий плеоцитоз лимфоцитарного характера. На ЭКГ выявляют преобладание неспецифической медленноволновой активности. Дифференциальный диагноз проводят с амавротической идиотией, лейкодистрофиями, миоклонус-эпилепсией, энцефаломиелитом и инфекционно-аллергического генеза.
В качестве средств симптоматической терапии, способствующих некоторой стабилизации состояния, рекомендуется применение тиамина-хлорида, кокарбоксилазы, пиридоксина-хлорида, кортикостероидных гормонов. При наличии в семье ребенка с Л.б. необходима медико-генетическая консультация.
При микроскопическом исследовании — спонгиоформное поражение, представляющее собой кистозные полости и гибель нейронов, демиелинизация и сосудистая пролиферация. Характерно повышение уровня лактата в сыворотке крови. Прогноз в целом неблагоприятный, однако у некоторых пациентов наблюдается длительная ремиссия.
! Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку - так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
+7 (925) 50 254 50 – срочное лечение в Италии