Русский Медицинский Сервер / Лечение в Италии / Центр лечения редких заболеваний в Милане / Синдром Марфана - лечение в Италии
Синдром Марфана - лечение в Италии
Синдром Марфана относится к моногенным болезням соединительной ткани - группе различных по происхождению нозологических форм, которые объединяют наследственные нарушения обмена соединительной ткани. Большая часть этой патологии обусловлена нарушением ферментных систем, контролирующих синтез структурных белков из которых происходит синтез соединительной ткани. Почти все эти болезни, входящие в состав синдрома, приводят к тяжелым инвалидизирующим расстройствам.
Синдром Марфана впервые описан Вильямсом в 1876 г. Свое название заболевание получило от французского педиатра Марфана, наблюдавшего девочку с характерным симптомокомплексом болезни 20 лет спустя. Существует интересный факт, что первая девушка модель - Лесли Хорнби, которая послужила прототипом образа всех моделей, имела синдром Марфана. Так, установлено, что ряд всемирно известных людей страдали синдромом Марфана, среди них следует упомянуть президента США А. Линкольна и великого скрипача Паганини.
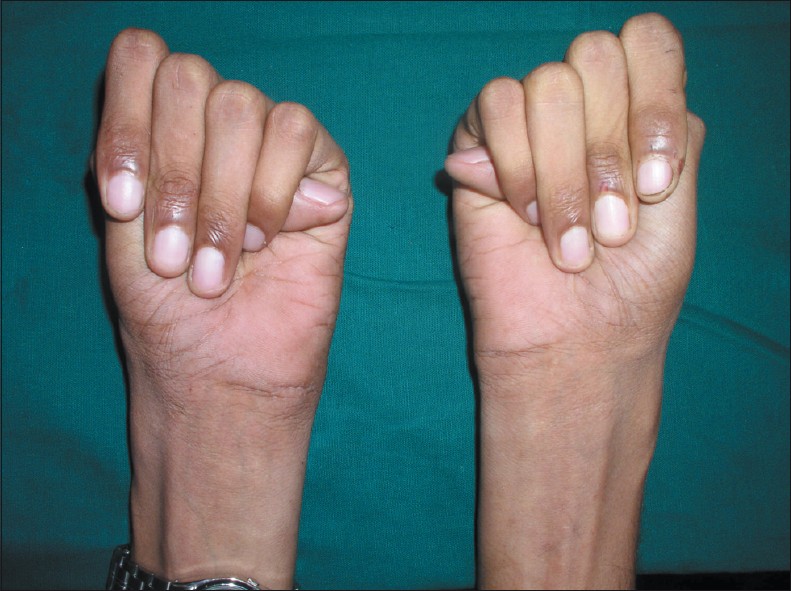
Клиническая картина заболевания характеризуется поражением многих жизненно важных органов и систем: опорно-двигательного аппарата, сердечно-сосудистой системы, органов дыхания и зрения, ЦНС.
Так, среди конституциональных особенностей и нарушений скелета наиболее часто встречаются долихопластический (астенический) тип, высокий рост (как правило, выше 180см) при выраженном дефиците массы тела (обычно ниже 50 кг.), арахнодактилия («паучьи» пальцы) кистей и стоп, кифосколиоз, воронкообразная или килевидная деформации грудной клетки, плоскостопие, узкий лицевой скелет, «готическое» небо. Не обязательно у одного человека встречаются все эти признаки, это лишь наиболее распространееные
Типичны также антимонголоидный разрез глаз, «крупный» нос, большие низкорасположенные ушные раковины, «птичье» выражение лица. У новорожденных из перечисленных особенностей скелета, как правило, выявляются только долихопластический тип и арахнодактилия. Остальные симптомы формируются в более поздние периоды развития (обычно в течение первых семи лет жизни ребенка).
Поражение сердца и сосудов - один из кардинальных признаков синдрома Марфана. Наиболее типичны среди них - пролапс митрального клапана и аневризма аорты. Сердечно-сосудистые нарушения регистрируются уже на первом-втором годах жизни ребенка, при этом отмечается постепенное увеличение диаметра аорты, достигающее критических размеров (до 6 см и более), чаще в возрасте от 16 до 45 лет.
Грозным осложнением аневризмы аорты является расслоение ее стенок, которое может быстро прогрессировать, захватывая всю длину аорты и отходящие от нее сосуды. Такие осложнения, как правило, заканчиваются летально.
Бронхолегочная система также вовлекается в патологический процесс при синдроме Марфана. Предпосылкой для этого являются механическое сдавление дыхательных путей при деформациях грудной клетки и изменения соединительнотканных структур легочной ткани. Нарушения органов дыхания в виде спонтанного пневмоторакса, легочной эмфиземы, инфаркта легкого встречаются с частотой от 10 до 75%. Наряду с этим, имеются сведения о врожденном недоразвитии одной из долей легкого, поликистозе легких, врожденной буллезной эмфиземе, двусторонних бронхоэктазах.
Диагноз синдрома Марфана основывается на семейном анамнезе, наличии у больного типичных диагностических признаков по результатам физикального осмотра, ЭКГ и ЭхоКГ, офтальмологического и рентгенологического обследования, молекулярно-генетического анализа и лабораторных исследований.
За диагностические критерии синдрома Марфана берутся характерные изменения в различных системах и органах; главными (большими) из них считаются: дилатация корня/расслоение восходящей части аорты, эктопия хрусталика и эктазия твердой мозговой оболочки; килевидная/воронкообразная деформация грудной клетки, требующая хирургического лечения; отношение длины верхнего сегмента тела к нижнему < 0,86 или размаха рук к росту > 1,05; сколиоз (> 20?) или спондилолистез; ограничение разгибания в локтевом суставе (<170о); плоскостопие; протрузия вертлужной впадины. Остальные проявления относятся к малым критериям, а генетические (семейные) признаки – к дополнительным. Для установления диагноза синдрома Марфана необходимо наличие минимум по 1-му большому критерию в двух системах органов и 1-го малого - в третьей; в скелетной системе – присутствие минимум 4-х больших.
Также применяются фенотипические диагностические тесты, определяющие соотношение кисть/рост (при синдроме Марфана > 11%); длину среднего пальца (> 10 см); индекс телосложения Варги – (масса тела, г/(рост, см)x2 – возраст, годы/100, должно быть <1,5); тест большого пальца на арахнодактилию, тест охвата запястья.
ЭКГ при синдроме Марфана позволяет определить нарушение ритма сердца, выраженную гипертрофию миокарда; ЭхоКГ - обнаружить клапанную регургитацию, увеличение размеров левого желудочка, пролапс митрального клапана, разрывы хорд, дилатацию аорты. На рентгенографии грудной клетки можно увидеть расширение корня и дуги аорты, увеличение размеров сердца; на КТ и МРТ сердца и сосудов - выявить дилатацию и аневризмы аорты.
Лечение и дальнейшее наблюдение пациентов с синдромом Марфана должно осуществляться группой специалистов: офтальмологом, кардиологом, кардиохирургом, ортопедом, генетиком, терапевтом.
Лечение больных с синдромом Марфана направлено на профилактику прогрессирования заболевания и развития осложнений, в первую очередь в сердечно-сосудистой системе. При диаметре аорты до 4 см назначаются ?-адреноблокаторы, антагонисты кальция или ингибиторы АПФ. Хирургическое лечение проводится при недостаточности клапанов сердца, пролапсе митрального клапана, значительном расширении (>5 см) восходящей части и расслоении аорты. Реконструктивные операции на аорте при синдроме Марфана, имеют высокий процент послеоперационной 5-ти и 10-ти летней выживаемости.
При необходимости выполняют протезирование митрального клапана. У беременных с синдромом Марфана и выраженной сердечно-сосудистой патологией проводят досрочное оперативное родоразрешение путем кесарева сечения. С целью профилактика инфекционного эндокардита и тромбозов после операционных вмешательств назначаются антибиотики и антикоагулянты.
При синдроме Марфана проводится коррекция зрения с помощью подбора очков и контактных линз, при необходимости – лазерное или хирургическое лечение катаракты, глаукомы, удаление смещенного хрусталика с имплантацией искусственного. При выраженных скелетных нарушениях может потребоваться хирургическая стабилизация позвоночника, торакопластика, эндопротезирование тазобедренных суставов. Применяются также патогенетическая коллагеннормализующая терапия, метаболическая и витаминотерапия.
! Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку - так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
+7 (925) 50 254 50 – срочное лечение в Италии