Русский Медицинский Сервер / Лечение в Италии / Центр лечения редких заболеваний в Милане / Синдром Секкеля - лечение в Италии
Синдром Секкеля - лечение в Италии
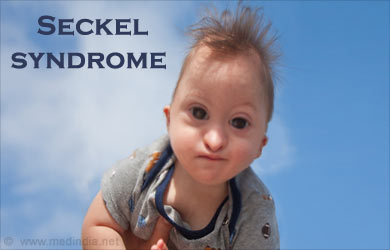
Синдром Секкеля (синонимы: карликовость с «птицеголовостью», примордиальная карликовость с микроцефалией) - редкое наследственное заболевание, характеризующееся пре- и постнатальным отставанием в росте, микроцефалией с задержкой психического развития и характерным изменением лица, напоминающим «птицеголовость».
Заболевание, по всей вероятности, было известно давно, так как термины «птицеголовая карликовость» и «наноцефалия» принадлежат Н.Зеске в 1960 г. опубликовал статью, в которой привел результаты обследования двух собственных пациентов и литературные данные о 24 больных, имевших сходные клинические проявления
Синдром Секкеля - редкий синдром, характеризующийся тяжелой низкорослостью, низким весом при рождении, очень маленькой головой (микроцефалия), большим лбом, большими глазами, низкими ушами, большим носом и небольшим подбородком. Дефекты костей рук и ног, вывихи локтевых суставов и бедер, неспособность выпрямлять колени и крипторхизм, также часто встречаются у детей с этим синдромом.
Хромосомная нестабильность и недопроизводство всех типов клеток крови (панцитопения) отмечаются у некоторых пациентов. Это заболевание является генетическим. Оно наследуется по аутосомно-рецессивному типу. Синдром Секкеля является неоднородным генетическим заболеванием, которое может быть связано с мутациями в генах, расположенных на хромосомах 3 и 18.
Следует отличать синдрос Секкеля от другиз заболеваний с низкорослостью. Для этого проводится дифференциальная диагностика при характерных дополнительных симптомах.
Низкая масса тела при рождении в случае доношенной беременности, внутриутробная задержка роста. Этот частый симптом свидетельствует о наличии генетически обусловленного снижения потенции роста тканей, недостаточности плаценты или же нарушений роста во внутриутробном периоде токсического или воспалительного генеза. Если отсутствует дополнительная симптоматика и вместе с тем имеющуюся клиническую картину нельзя отнести к какому-либо определенному заболеванию, говорят о примордиальной низкорослости. При отсутствии других симптомов последняя описывается как семейное заболевание.
В спорадических случаях следует думать о необратимом внутриутробном повреждении редупликационного потенциала клеток неизвестным патогенным фактором. При чисто примордиальной низкорослости дифференцировка скелета соответствует хронологическому возрасту.
- а) Примордиальная низкорослость: синдром Рассела - Сильвера.
- б) Синдром Секкеля: гетерогенная группа примордиальных карликов с микроцефалией, большим носом, скошенным подбородком и интеллектуальным недоразвитием.
- в) Прогерия (синдром Гатчинсона-Джилфорда): замедление роста и развитие начиная со 2-3-го года жизни, преждевременное старение, атрофия кожи, выпадение волос.
- г) Синдром Блума: телеангиэктатическая эритема щек, долихоцефалия.
- д) Синдром Корнелии де Ланге; умственное недоразвитие, густые, сросшиеся над переносицей брови, микроцефалия, усиленное оволосение тела.
- е) Лепрехаунизм (синдром Донохуа): отсутствие или недоразвитие подкожной жировой клетчатки, гипертрофия клитора или полового члена, усиленное оволосение.
- ж) Синдром Фанкони: панцитопения, гипоплазия I пальца руки или нарушение его закладки, пигментные аномалии.
- з) Семейная, или полигенная, низкорослость, при которой в большинстве случаев наблюдается только легкая задержка роста во внутриутробном периоде.
- и) Трисомия 18.
Очень короткие конечности (увеличенное отношение верхнего сегмента к нижнему). К этой группе прежде всего относятся заболевания скелета, обусловленные поражением эпифизов и метафизов. При дифференциальной диагностике обязательно следует подумать об атиреозе или тяжелом гипотиреозе (особенно учитывая возможности их лечения). Фосфатный диабет также может сопровождаться легкой диспропорциональной низкорослостью с преобладанием верхнего сегмента. Пропорции могут существенно меняться в процессе роста. Здесь представлены сведения о пациентах старшего детского и юношеского возраста в соответствии с обобщением Спрангера.
- а) Ахондроплазия (хондродистрофия): относительно большая голова с провалившимся основанием носа, особенное укорочение предплечий и голеней, кифоз, типичные рентгенологические находки.
- б) Гипохондроплазия: слабо выраженная диспропорциональная низкорослость, переразгибание суставов, люмбальный лордоз, типичные рентгенологические данные.
- в) Псевдоахондроплазия: пропорции, как и при ахондроплазии, но с нормальным формированием мозгового и лицевого черепа, относительно позднее по сравнению с ахондроплазией проявление заболевания, отсутствие типичных рентгенологических симптомов со стороны таза.
- г) Синдром Эллиса-Ван-Кревельда: порок сердца, дистрофия ногтей и пальцев стоп, эписпадия.
- д) Синдром Маркезани: изменения хрусталиков, как при синдроме Марфана (дрожание хрусталиков, может быть шаровидный хрусталик), брахидактилия, возможны легкие контрактуры.
- е) Метафизарная хондродисплазия, тип Шмидта: укорочение трубчатых костей (больше проксимальных), грибовидно вздутые эпифизы с размытыми очертаниями.
- ж) Метафизарная хондродисплазия, тип Мак-Кусика: редкие тонкие волосы, брови и ресницы, переразгибание суставов, типичная рентгенологическая картина.
- з) Метафизарная хондродисплазия, тип Енсена: утолщенные суставы с костным ограничением подвижности, формирование косолапости стоп, типичная рентгенологическая картина.
- и) Дисхондростеоз (синдром Лери - Вейля): укорочение предплечья и лучевой кости по отношению к локтевой, дорсальный, поддающийся редрессации подвывих дистального конца локтевой кости (деформация Маделунга).
- к) Пикнодизостоз: короткие концевые фаланги, большой мозговой череп, большие роднички (которые могут быть открытыми до зрелого возраста), спонтанные переломы.
- л) Карликовость с микромелией: гипоплазированные локтевая и малоберцовая кости, а также нижняя челюсть.
- м) Карликовость с микромелией: характерная "треугольная" деформация большеберцовой кости.
- н) Псевдогипопаратиреоз: ожирение, обращающее на себя внимание круглое лицо, укороченные пальцы рук, изменения метаболизма.
- о) Псевдопсевдогипопаратиреоз.
! Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку - так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
+7 (925) 50 254 50 – срочное лечение в Италии