Фертильность у женщин с синдромом Шерешевского-Тернера.
|
Словарь
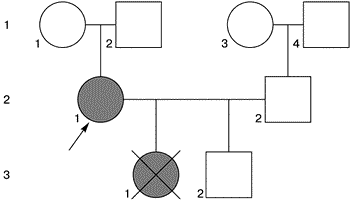
Кариотип – полный набор хромосом диплоидной клетки (кариотип человека в норме 46,ХХ или 46,ХY). Фенотип – совокупность признаков организма, контролируемый определенным генотипом. Стигмы – фенотипические отклонения от нормального развития. Фертильность – детородная функция живого организма. Гоносомы – половые хромосомы это Х- и Y-хромосома. Моносомия – утрата целой хромосомы. Стреки – соединительнотканные тяжи. Центромера – органоид, располагающийся в районе первичной перетяжки хромосомы. Она делит хромосому на два плеча. Теломера – концевой участок хромосомы. Специализированная структура, обеспечивающая стабильность линейной молекулы ДНК. Делеция – утрата участка хромосомы. Дисгенезия –нарушение формирования половой железы и вторичных признаков пола. Полная или частичная потеря одной хромосомы Х ведет большинстве случаев к абсолютному бесплодию. Но в 1960 г. F.Bahner и соавт. [2] впервые сообщили о фертильности у пациентки с синдромом Шерешевского–Тернера и с кариотипом 45,Х0. С этого времени описано более 100 беременностей у женщин с синдромом Шерешевского–Тернера. При этом беременные имели в большинстве гоносомальный мозаицизм между одной клеточной линией с различной аберрантной хромосомой Х или моносомией Х и одной нормальной линией клеток 46,ХХ, причем отношения обеих клеточных линий были различными. Дети матерей с мозаицизмом, например 46,ХХ/45,Х, имели как нормальный кариотип [13], так и аберрантный гоносомальный клон клеток [3]. Эти данные говорят, что потеря одной полной хромосомы Х или какой-то одной части хромосомы Х, не в каждом случае должна сопровождаться бесплодием [3, 14]. Приводим наши наблюдения фертильности у женщин с бесплодием, невынашиванием, имеющих детей с врожденными пороками развития в анамнезе, с мозаичным кариотипом 46,ХХ/45,Х. Больная К. обратилась в медико-генетический центр 26.07.95 с целью генетического обследования и решения вопроса о прогнозе будущего потомства. Из акушерского анамнеза выяснено, что 29.08.95 в сроке 26–27 нед была прервана беременность. Причиной для прерывания беременности послужило заключение ультразвукового исследования (УЗИ), которое выявило множественные врожденные пороки развития плода: укорочение нижних конечностей, двухкамерное сердце, гидроцефалия. После прерывания беременности у плода мужского пола, массой 0,925 кг, были установлены все выявленные на УЗИ пороки развития. Из анамнеза женщины: менархе в 12 лет, месячные спонтанные, регулярные, через 24 дня по 3 дня, безболезненные, умеренные. Половая жизнь с 21 года. Беременность наступила спонтанно в течение первого года регулярной половой жизни и закончилась 29.08.95 в сроке 26–27 нед прерыванием беременности по медицинским показаниям. Генеалогический анамнез семьи пробанда представлен на рис. 1.
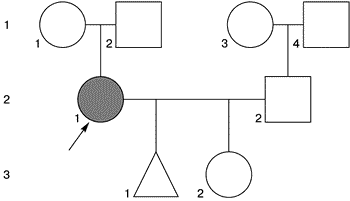
Данные обследования. Женщина правильного телосложения, рост 160 см, масса 56 кг. Вторичные половые признаки развиты удовлетворительно. Отмечается неправильный прикус, неправильный рост зубов нижней и верхней челюсти, высокое небо. Значительных стигмальных отклонений не выявлено. При хромосомном анализе у мужа нормальный мужской кариотип 46,XY. В кариотипе пробанда выявлена патологический клон клеток с моносомной X-конституцией – 11% (кариотип 46,XX/45,X – 11%). 23.06.97 больная вновь обратилась в медико-генетический центр по поводу прогноза потомства в сроке беременности 30 нед. При УЗИ патологии со стороны внутриутробного плода не выявлено. Гормональный скрининг и инвазивная пренатальная диагностика не проводились в связи с обращением в центр в позднем сроке беременности. Беременность пролонгировалась под наблюдением врача женской консультации. 14.10.97 срочные роды без осложнений в срок 39 нед беременности. Родился здоровый мальчик массой 4450 г и длиной тела 54 см. Из родильного дома был выписан домой на 8-е сутки жизни. При осмотре ребенка в медико-генетическом центре патологических отклонений в фенотипе не выявлено. Кариотипический анализ лимфоцитов периферической крови ребенка показал нормальный мужской хромосомный набор 46,XY, без числовых и структурных перестроек. Больная Б. 22 года, медицинская сестра. Обратилась в медико-генетический центр 5.09.95 с жалобами на отсутствие беременности в течение 1 года после замужества. Состоит в первом неродственном браке. Мужу 22 года. Из анамнеза выяснено: менархе в 12 лет, месячные нерегулярные: интервал от 32 до 44 дней, менструации по 4 дня, очень болезненные. Половая жизнь с 21 года без контрацепции. Генеалогический анамнез представлен на рис. 2.
Данные обследования. Правильного телосложения, рост 154 см, масса 56 кг. При осмотре: диспластичные ушные раковины, выступающий фильтр, диастема, высокое небо, низкий рост волос на шее, широкий плечевой пояс, дисплазия ногтевых пластин. Половая формула Ме+ Ма2 Ах3 Рв3. УЗИ внутренних гениталий (на 11-й день цикла): матка размерами 47' 30' 38 мм, грушевидной формы, шейка матки 25 мм, эндометрий 7 мм, правый яичник 47' 25' 25 мм, левый – 46' 26' 26 мм, содержит множество жидкостных включений в диаметре до 5–6 мм. Гормоны крови (7-й день цикла): тестостерон 11,3 нм/л, пролактин 446 нг/мл, ЛГ 19,7 мМЕ/мл, ФСГ 5,5 мМЕ/мл, ТТГ 2,8 МЕ/л. Кариотип – 46,ХХ/45,Х – 15%, кариотип мужа 46,XY, без числовых и структурных перестроек. Через 7 мес после первого обращения в медико-генетический центр спонтанно наступила беременность, которая самопроизвольно прервалась в сроке 5–6 нед. Через 2 года после индукции овуляции клостильбегитом в дозе 50 мг в день с 5-го по 9-й день цикла наступила беременность. При медико-генетическом консультировании рекомендовано проведение комплекса мероприятий по пренатальной диагностике. При гормональном исследовании крови по программе пренатальной диагностики "АЛЬФА" в срок беременности 16–17 нед от 8.02.99: концентрация a-фетопротеина – 37,6 МЕ/мл – 0,95 МОМ; ХГ – 47 340 МЕ/л – 1,64 МОМ; Е3 – 33,6 нмоль/л – 1,05 МОМ. Результат скрининга: риск рождения ребенка с болезнью Дауна 1:1 300; риск рождения ребенка с дефектом невральной трубки – 1:12 000. При УЗИ плода в срок беременности 23 и 34 нед патологии не выявлено. С целью исключения хромосомной патологии плода 19.01.99 беременной был произведен трансабдоминальный кордоцентез. Анализ кариотипирования лимфоцитов крови плода показал нормальный женский кариотип 46,ХХ, без числовых и структурных перестроек. Учитывая наличие нормального кариотипа у плода, женщина пролонгировала беременность. Роды произошли в срок беременности 38 нед. Родилась здоровая девочка массой 3300 г, длиной 54 см. Обсуждение По современным представлениям, гонады эмбриона с кариотипом 45,Х дегенерируют в позднем фетальном периоде или в раннем периоде новорожденности и представляют собой типичные гонады тяжи [1]. Период до полной атрезии всех фолликулов может длиться до 20 или 30 лет, поэтому существование активных фолликулов при дисгенезии гонад возможно и у взрослых индивидуумов. Клиническая картина зависит от структуры дисгенетичной гонады. Возможен спонтанный пубертат как в представленных случаях и/или ранняя вторичная аменорея. Так, A.M. Pasquino и соавт. сообщают о 522 девочках с синдромом Шерешевского–Тернера, старше 12 лет, не имеющих отчетливых признаков заболевания, с кариотипом 45,Х и 45,X/46,XX [12]. По данным этих авторов, у 84 больных был спонтанный пубертат с менархе в 13 лет. При этом костный возраст в среднем составил 12,9 года. Регулярный менструальный цикл сохранялся у 30 больных в среднем 9,2 года после менархе. Вторичная аменорея развилась в среднем через 1,6 года. У некоторых менструации приобрели нерегулярный характер через 9 мес. У 3 больных наступила беременность, которая закончилась в одном случае рождением здорового ребенка, в другом – рождением ребенка с синдромом Тернера и в третьем – рождением ребенка с врожденными пороками развития. C.Huseman [7] сообщает даже о случае с преждевременным пубертатом, с менархе в 10-месячном возрасте при кариотипе 45,Х/46,ХХ. Возможно имело место персистирование функционально полноценного фолликула при наличии клеточной линии с нормальным кариотипом. Отметим, что в представленных случаях у больных был мозаичный кариотип с преобладанием нормальной ХХ-клеточной линии в лимфоцитах периферической крови. Нормальное развитие яичников во время фетального периода с наступающей после рождения регрессией описано у девочек с хромосомной конституцией 46,X,i (Xq) [6]. A.Farabosco и соавт. указывают, что хромосомные участки Xq13- больше q21 и Хq21- больше q26 ответственны за развитие фертильных гонад. Авторы приводят данные о фертильности женщин с гетерозиготным дефицитом сегмета Хр ter- больше p21 [5]. Возможно, гены, необходимые для поддержания функциональной способности ооцитов, локализованы между Хp21 и центромерой [6]. Около 5–11% всех фертильных женщин с синдромом Шерешевского–Тернера имеют кольцевую хромосому Х с кариотипическим мозаицизмом 45Х/46,Х,r(X). Происхождение радиарной хромосомы обусловлено делецией обеих теломер или теломерных сегментов с присоединенным слиянием мест переломов хромосом X или Y [8]. E.Bannier и соавт. [3] сообщают о случае передачи кольцевой хромосомы Х от фергильной матери к дочери. Современные методы определения происхождения подобной хромосомы (неауторадиографическая гибридизация in situ биотинированных Х (qSV2X5)- и Y (Y84)-специфических для центромер зондов) позволяют четко и быстро идентифицировать кольцо Х-происхождения [4] у матери и потомка. Описан также случай синдрома Шерешевского–Тернера у пробанда с кариотипом 45,Х/46,Х, del (X) (q21.2-q ter)/46,X, r(X), имеющего функциональную дисомию и изодисомию материнского происхождения [9], что указывает на возможность передачи аберрантной хромосомы от фертильнои матери к потомству. Ребенок от женщины с мозаичным вариантом синдрома Шерешевского–Тернера имел в нашем случае нормальный кариотип. Это позволяет считать, что созревшая яйцеклетка должна содержать неаберрантную хромосому Х. Эту мысль подтверждает A.Novac и соавт. [11] и A.Mori и соавт. [10], которые нашли разную патологию в хромосомном статусе периферической крови, кожных фибробластов, а также левой и правой гонад. Таким образом, кариотип, определенный в лимфоцитах периферической крови, не в каждом случае определяет ситуацию в гонадах. Заключение Наличие у женщин кариотипа с патологическим клоном клеток (с мозаичным или моносомным вариантом синдрома Шерешевского–Тернера) не исключает наступления у них спонтанных, неиндуцированных беременностей и рождения здоровых детей. Разнообразие в экспрессии клинической картину синдрома Шерешевского–Тернера удивительно. Оно не характеризуется только тернеровскими стигмами и степенью гонадальной дисгенезии. При синдроме Шерешевского–Тернера спонтанные менструации при патологической гоносомной конституции встречаются чаще, чем нормальная фертильность. Т.А. Соколова, Г.Г. Шупта, М.П. Корф Красноярский межрегиональный медико-генетический центр Литература 1. Кирпатовский И.Д.,„ Голубева И.В. Патология и коррекция пола. М. Изд Российского университета дружбы народов 1992; 21–23. 2. Bahner F.,„ Schwarz G., Heinz H., Walter K. Acta Endocrinol 1960; 35: 397. 3. Bannier E., Gedschold J., Missbach D„., Gotze A. und and. Z Klin Med 1990; 45: Heft. 9: 761–763. 4. Crolla J.A., Llerena J. Hum Genet 1988; 81: 1: 81–84. 5. Farabosco A., Giorgio L., Formics A. et al. Clin Genet 1979; 22: 11–16. 6. Hodgson S.D., Chin D.,„ Polani P. Hum Genet 1981; 58: 176–178. 7. Huseman C.A. J Pediat 1982; 102: 892–894. 8. Jani Mihir M., Torchia Beth S., Pai G. et al. Genomics 1995; 27: 1: 182–188. 9. Migeon R., Jeppesen P., Torchia Bath S. et al. Amer J Hum Genet 1996; 58: 1: 161–170. 10. Mori A„., Jamakita N., Ho J. Folia Endocrinol jap 1990; 66: 3: 175–181. 11. Novac Angelina Z.,„ Kakai George K., Popovic Vera P. Hum Genet 1995; 95: 3: 293–298. 12. Pasquino A.M., Passeri F., Pucarelli I. J Of Clin Endocrinolog and Metab 1997; 82: 6: 1810–1813. 13. Tuschy U., Birgit John, Wevner W. Zbl Gynakol 1985; 107: 1384–1387. 14. Tyrkis M., Hoffman W.H„., Кraemer-Flynn K.M. Clin Genet 1988; 35: 2: 111–115. |