Инсулинорезистентность у пациенток с синдромом поликистозных яичников (обзор литературы).
|
Синдрому поликистозных яичников (СПКЯ) в настоящее время уделяется повышенное внимание. Частота данной патологии среди гинекологических заболеваний значительна – от 1,8 до 11%. Несмотря на большое количество исследований в этой области, отдельные звенья патогенеза при СПКЯ остаются не до конца ясны.
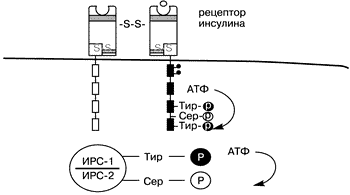
В настоящем обзоре представлены данные зарубежной литературы о взаимосвязи СПКЯ, инсулина и факторов роста. С тех пор как G. Burghen и соавт. [8] в 1980 г. показали, что СПКЯ сочетается с гиперинсулинемией, стал очевиден факт развития при данном синдроме серьезных метаболических и репродуктивных нарушений. Изучению связи между инсулином и функцией гонад посвящены многочисленные исследования [4, 22, 25, 62, 63]. Впервые на взаимосвязь нарушений углеводного обмена и гиперандрогенемии указали С. Achard и J.Thiers в 1921 г. [2]. Это сочетание было названо “диабетом бородатых женщин”. R. Kierland и соавт. в 1947 г. обнаружили у женщин с гиперандрогенией и сахарным диабетом специфическое нарушение пигментации кожи, названное acanthosis nigricans, [48]. В 1968 г. J.Brown и R. Winkelmann [7] предположили, что данное состояние является инсулинорезистентным сахарным диабетом. Обнаружение этого заболевания у сестер-близнецов в сочетании с признаками акромегалии [15] позволило предположить наличие генетической основы данной патологии [5]. Следует отметить, что сочетание гиперандрогении, инсулинорезистентного сахарного диабета и acanthosis nigricans наблюдается и при некоторых других синдромах, в частности, при липоатрофическом диабетическом синдроме, лепрекаунизме, синдроме Рабсон–Мендельшталла, синдроме типов А и В [18, 25, 73]. Синдром типа A, который встречается у девочек-подростков, был описан С.Kahn и соавт. [48] в 1976 г. [44]. При этом синдроме у девочек наблюдалась выраженная инсулинорезистентность в сочетании с сахарным диабетом, acanthosis nigricans, а также имелись грубые признаки вирилизации (увеличение мышечной массы, клиторомегалия, временная аллопеция, снижение тембра голоса). У женщин в постменопаузе авторы также наблюдали выраженную инсулинорезистентность с acanthosis nigricans и аутоиммунными нарушениями. Данное состояние было названо синдромом типа В, в основе которого, как предполагается, лежит образование эндогенных антител к инсулиновым рецепторам [31, 58]. В дальнейших исследованиях было доказано, что причинами лепрекаунизма, синдрома Рабсон–Мендельшталла и синдрома типа А являются мутации гена инсулинового рецептора [58, 73]. В 1980 г. G.Burghen и соавт. [8] на основании выявления у женщин с СПКЯ и признаками гиперандрогенемии, гипресулинеммии как базальной, так и глюкозостимулированной (в отличие от женщин с идентичной массой тела из группы контроля) предположили, что в основе этого явления лежит инсулинорезистентность. Они также установили наличие положительной корреляции между уровнем инсулина и уровнем андрогенов, что, как полагают авторы, может иметь этиологическое значение. В середине 80-х годов несколько групп исследователей сообщили о том, что acanthosis nigricans часто встречается у полных женщин с гиперандрогенемией [26, 30, 61, 69]. В сравнении с контрольной группой, идентичной по возрасту и массе, у этих пациенток имела место гиперинсулинемия как натощак, так и после проведения теста на толерантность к глюкозе. Наличие же гиперинсулинемии у женщин с СПКЯ не зависит от массы тела, что подтверждено многочисленными исследованиями [10, 59, 68]. Однако интолерантность к глюкозе все же наблюдается чаще у полных пациенток с СПКЯ по сравнению с контролем (30% против 10%), тогда как для худых пациенток с СПКЯ это нехарактерно [21, 64]. В популяционных исследованиях показано, что 16–80% женщин с СПКЯ имеют избыточную массу тела [20, 23, 64]. Именно у данной группы женщин чаще всего обнаруживается acanthosis nigricans [23, 24, 26, 61, 69]. По данным J.Holte [38], инсулинорезистентность у больных с СПКЯ наблюдается только при высоких значениях массоростового индекса (МРИ), так как у женщин со сниженным и нормальным МРИ имеет место так называемая “ранняя ” гиперинсулинемия. Под данным термином подразумевается увеличенный ранний инсулиновый ответ на нагрузку глюкозой, не связанный с инсулинорезистентностью [38]. По данным A.Dunaif и соавт. [26], у пациенток с СПКЯ и инсулинорезистентностью в яичниках помимо типичных для поликистоза морфологических изменений отмечается выраженный гипертекоз стромы яичников со значительным распространением островков лютеинизированных текаклеток в строме яичника, вследствие чего у них отмечается более выраженная гиперандрогения [ 41] . Однако P.Hughesdon [42] показал, что при тщательном исследовании яичников у женщин с СПКЯ также можно обнаружить маленькие островки гипертекоза. Но у инсулинорезистентных женщин с СПКЯ эти морфологические изменения более выражены, так как гиперинсулинемия оказывает влияние как на морфологию, так и на функцию яичников [26]. Эта гипотеза была подтверждена дальнейшими исследованиями [55], в которых была установлена положительная корреляция между гиперинсулинемией и гипертекозом овариальной стромы. Однако быстрое прогрессирование симптомов андрогенемии и/или истинной вирилизации – редкое явление при СПКЯ [32, 35, 79]. Для лучшего понимания связи СПКЯ с инсулинорезистентностью рассмотрим подробно структуру инсулинового рецептора и механизм действия инсулина. Клеточные и молекулярные механизмы инсулинорезистентности Инсулин действует на клетку путем связывания с рецептором, находящимся на поверхности клетки [11, 42, 43]. Инсулиновый рецептор – гетеротетрамер, состоящий из двух a,b-димеров, связанных дисульфидными мостиками [46] (рис. 1). Каждый a ,b-димер является продуктом одного гена (29, 75). a -Субъединица расположена экстрацеллюлярно и содержит связывающий домен, тогда как b-субъединица перекрывает мембрану. Цитоплазматическая часть b-субъединицы содержит протеинтирозинкиназу, которая активируется путем лигандопосредованного аутофосфорилирования на специфический тирозиновый остаток [47] (см. рис. 1).
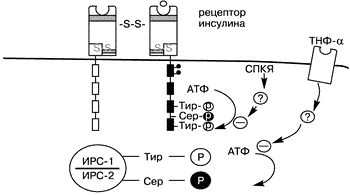
Инсулиновый рецептор относится к семейству протеинтирозинкиназных рецепторов, в которое входит и рецептор к инсулиноподобному фактору роста (ИПФР-1). Инсулиновый и ИПФР-1 рецепторы обладают существенной структурной гомологичностью К данному семейству относятся и рецепторы к эпидермальному фактору роста и колониестимулирующий фактор роста [1, 76]. Онкогенные продукты также являются протеинами тирозинкиназы [9, 75]. Цитоплазматическая часть b-субъединицы содержит протеинтирозинкиназу, которая активизируется путем лигандопосредованного аутофосфорилирования (вероятно, через конйормационные изменения) на специфический тирозиновый остаток [13, 33, 47, 67] (см. рис. 1). Активированный инсулиновый рецептор тирозинфосфорилирует внутриклеточные субстраты и запускает сигнал трансдукции [11]. Последнее время удалось выявить и охарактеризовать несколько субстратов, в частности субстрат инсулинового рецептора-1 (СИР-1), который используется как стыкующая молекула для сигнальных и адапторных молекул [54, 70]. Достаточно недавно был идентифицирован другой субстрат – субстрат инсулинового рецептора-2 (СИР-2) (60, 71). В результате происходящих каскадных механизмов запускается процесс инсулинопосредованного транспорта глюкозы. Известно, что тирозинаутофосфорилирование усиливает активность рецепторов тирозинкиназы, тогда как серинфосфорилирование подавляет ее [42]. Сделано предположение, что опорследованное протеинкиназой C серинфосфорилирование инсулинового рецептора приводит к инсулинорезистентности, индуцированной гипергликемией [49, 53]. При исследовании фибробластов у женщин с СПКЯ, A.Dunaif и соавт. приблизительно в половине случаев отметили снижение аутофосфорилирования инсулиновых рецепторов [27]. Учитывая, что серинфосфорилирование инсулинового рецептора ингибирует активность тирозинкиназы рецептора, что показано также во внеклеточных системах и in vivo, подобный дефект на ранних стадиях передачи инсулинового сигнала может быть причиной инсулинорезистентности у данной группы женщин с СПКЯ [6, 12, 19, 28, 45, 72]. Отсутствие нарушений аутофосфорилирования инсулинового рецептора у другой половины женщин с СПКЯ, вероятно, объясняется нарушениями на более поздних стадиях передачи инсулинового сигнала [28, 42]. Из сказанного выше следует, что имеются некие серинфосфолирирующий фактор, которые приводят к нарушению процесса фосфорилирования инсулинового рецептора, что в свою очередь является причиной инсулинорезистентности, ассоциированной с СПКЯ [19] (рис.2). Подобными факторами могут являться серинтреонинкиназа или ингибитор серинтреонинфосфатазы [28, 36]. Также имеются данные, что серинфосфорилирование СИР-1 лежит в основе механизма инсулинорезистентности, медиатором которой выступает фактор некроза опухолей. Гликопротеин мембраны РС-1 также ингибирует активность киназы инсулинового рецептора, но без серинфосфорилирования [19]. По мнению Miller и соавт., те же или подобные им факторы вызывают серинфосфорилирование P450c17, который является ключевым ферментом, контролирующим биосинтез андрогенов как в яичниках, так и в надпочечниках [80]. Аналогично тому, что серинфосфорилирование инсулинового рецептора приводит к к инсулинорезистентности, серинфосфорилирование P450с17 приводит к гиперандрогенемии. Исходя из этого, наличием одного генетического дефекта можно объяснить связь между СПКЯ и инсулинорезистентностю [19]. Кандидатом на роль этого фактора может выступить протеинкиназа А, способность которой вызывать серинфосфорилирование инсулинового рецептора и P450c17 показана in vitro [80].
До сих пор спорным для исследователей остается вопрос о том, вызывает ли гиперинсулинемия гиперандрогению. В период пременопаузы синдром крайней инсулинорезистентности обычно ассоциирован у женщин с гиперандрогенемией [18, 73]. В основе клеточного механизма инсулинорезистентности при данном синдроме (типа В) лежит наличие антител, блокирующих связывание инсулина с его рецепторами. К другим причинам развития инсулинорезистентности относится генетический дефект рецепторов, вследствие чего уменьшается их число или подавляется функция рецептора (синдром типа A, лепрекаунизм) [18, 73]. Известно также, что in vitro инсулин напрямую влияет на стероидогенез, стимулируя выработку яичниками эстрогенов, андрогенов и прогестерона. Хотя данное действие инсулина частично проявляется и при его физиологических концентрациях, оно более выражено при высоких концентрациях [18, 22, 57]. Инсулиновые рецепторы распространены в организме повсеместно, однако их наличие в мембране незрелых яичников не означает, что инсулин играет физиологическую роль в регуляции стероидогенеза [42, 43]. Низкая же концентрация инсулина в фолликулярной жидкости свидетельствует скорее о гематогенном пути попадания в нее, чем об его локальной продукции [17]. В то же время ИПФР-1 продуцируется именно тканью яичника, и рецепторы к ИПФР-1 так же представлены в яичнике [65, 66]. Доказана структурная и функциональная гомологичность ИПФР-1 и инсулина, а также их рецепторов [16]. Благодаря этой особенности инсулин может связываться с лигандсвязывающим доменом рецептора ИПФР-1, повышая активность тирозинкиназы его b-субъединицы [16, 34, 75, 76]. Однако сродство рецептора ИПФР-1 к инсулину значительно меньше, чем к ИПФР-1, и наоборот [34]. Однако афинность варьирует в зависимости от типа ткани, и существуют рецепторы ИПФР-1, которые обладают равным сродством с МПФР-1 и инсулином. Их называют "атипическими" [37, 40], a, b - Димеры инсулина и рецептор ИПФР-1 могут соединяться и формировать гибридные гетеротетрамеры [50–52, 62]. Основными регуляторами активности ИПФР являются ИПФР-связывающие протеины (ИПФРСП). Инсулин снижает уровнеь продуцируемого печенью ИПФРСП-1, в результате чего ИПФР-1 становится более биодоступным [50]. С рецепторами других факторов роста (ИПФР-2, ЭФР, трансформирующий фактор роста- a,b, которые играют определенную роль в яичниковом стероидогенезе, инсулин непосредственно не может связываться. Однако известно, что рецепторы некоторых факторов роста, как ЭФР, также являются протеинкиназами [22, 46, 76]. Следовательно, возможно перекрестное взаимодействие между системой инсулин-ИПФР-1 и другими системами протеинкиназных факторов роста [3, 42]. Например, серинфосфорилирование рецептора ЭФР снижает активность тирозинкиназы [14, 74]. Инсулин в высоких концентрациях может мимикрировать действие ИПФР-1, благодаря связыванию с рецептором ИПФР-1 [22, 34, 50]. Это, вероятно, является одним из возможных механизмов развития инсулин-опосредованной гиперандрогенемии [4, 25, 63]. Кроме того, инсулин может оказывать действие на стероидогенез и непосредственно через свои рецепторы [77]. Эти механизмы имеют местои при инсулинорезистентных состояниях (77, 78) вследствие различной концентрации рецепторов в тканях или различной чувствительности рецепторов к инсулину. При применении у пациенток с СПКЯ препаратов, снижающих секрецию инсулина (диазоксид, соматостатин) либо повышающих чувствительность к инсулину (метформин, троглитазон), отмечено снижение концентрации андрогенов и повышение концентрации ПССГ [27, 56]. Поскольку у здоровых женщин инсулин не оказывает существенного воздействия на уровень циркулирующих андрогенов, можно предположить, что для подобного действия инсулина необходимо наличие предрасполагающих факторов. К ним следует отнести изменения в яичниках, характерные для СПКЯ (гиперплазия текаклеток). Таким образом, в настоящее время установлено, что инсулинорезистентность, которая часто сочетается с СПКЯ, в 50% случаев связана с избыточным серинфосфорилированием рецептора инсулина. Внешний фактор, скорее всего серинтрионинкиназа, является причиной этого нарушения. Серинфосфорилирование изменяет активность р450с17, ключевого регуляторного фермента биосинтеза андрогенов. Возможно, что у некоторых женщин с СПКЯ причиной возникновения инсулинорезистентности и гиперандрогении является один и тот же генетический дефект. В.А. Бурлев, Н.С. Аванесян, А.С. Гаспаров, Т. Нессен, Р. Хенриксен, Д. Кириловас Научный центр акушерства, гинекологии и перинатологии РАМН, Москва; Отделение акушерства и гинекологии университета г. Уппсала, Швеция Литература 1. Бурлев В.А., Гаспаров А.С., Аванесян Н.С., Волков Н.И., Стыгар Д.А. Факторы роста и их роль в регуляции репродуктивной функции у больных с синдромом поликистозных яичников. Пробл репрод 1998, 3: 17–25. 2. Achard C., Thiers J. Le virilisme pilaire et son association a I’insuffisance glycolytigue (diabete des femmes a barb). Bull Acad Natl Med 1921; 86: 51–64. 3. Ballotti R., Lammers R., Scimeca J. Intermolecular transphosphorylation between insulin receptors and EGF-insulin receptor chimerae. EMBO J 1989; 8: 3303–3309. 4. Barbieri R.L., Ryan K.J. Hyperandrogenism, insulin resistance, and acantosis nigricans syndrome: a common endocrinopathy with distinct pathophysiologic features. Am J Obstet Gynecol 1983; 147: 90. 5. Barnes N.D., Palumbo P.J., Hayles A.M., Folgar H. Insulin resistance, skin changes and virilization: a recessively inherited syndrome possibly due to pineal gland dysfunction. Diabetologia 1974; 10: 284–289. 6. Bollade G.E., Roth R.A., Beaudoin J., Mochly-Rosen D., Koshland Jr. D.E. Protein kinase C directly phosphorylates the insulin receptor in vitro and reduces its protein-tyrosine kinase activity. Proc Natl Acad Sci USA 1986; 83: 5822–5824. 7. Brown J., Winkelmann R.K. Acanthosis nigricans: a study of 90 cases. Medicine 1968; 47: 33–51. 8. Burghen G.A., Givens J.R., Kitabchi A.E. Correlation of hyperandrogenism with hyperinsulinism in polycystic ovarian disease. J Clin Endocrinol Metab 1980; 50: 113–116. 9. Cantley L.C., Auger K.R., Carpenter C., Duckworth B., Graziani A., Kapeller R., Soltoff S. Oncogenes and signal transduction. Cell 64: 281–302 [published erratum appears in Cell 1991 1991; May 31: 65(5): following 914]. 10. Chang R.J., Nakamura R.M., Judd H.L., Kaplan S.A. Insulin resistance in nonobese patients with polycystic ovarian disease. J Clin Endocrinol Metab 1983; 57: 356–359. 11. Cheatham B., Kahn C.R. Insulin action and the insulin signaling network. Endocr Rev 1995; 16: 117–142. 12. Chin J.E., Liu F., Roth R.A. Activation of protein kinase C inhibits insulin-stimulated tyrosine phosphorylation of insulin receptor substrate-1. Mol Endocrinol 1994; 8: 51–58. 13. Cobb M.H., Sang B.C., Gonzalez R., Goldsmith E., Ellis L. Autophosphorylation activates the soluble endoplasmic domain of the insulin receptor in an intermolecular reaction. J Biol Chem 1989; 264: 18701–18706. 14. Cochet C., Gill G.N., Meisenhelder J., Cooper J.A., Hunter T. C-kinase phosphorylates the epidermal growth factor receptor and reduces its epidermal growth factor-stimulated tyrosine protein kinase activity. J Biol Chem 1984; 259: 2553–2558. 15. Colle M., Doyard P., Chaussain J., Battin J., Job J. Acanthosis nigricans, hirsutisme et diabete insulin–resistant. Arch Fr Pediatr 1979; 36: 518–523. 16. Crowley W.F. New tools for pituitary-gonadal assessment. In: Filicori M, Flagmini C. (eds) The Ovary: Regulation, Dysfunction and Treatment. Elsevier, Amsterdam, 1996; 287–293. 17. Czech M.P. Structural and functional homologies in the receptors for insulin and the insulin-like growth factors. Ceii 1982; 31: 8–10. 18. Diamond M.P., Webster B.W., Carr R.K., Wentz A.C., Osteen K.G. Human follicular fluid insulin concentrations. J Clin Endocrinol Metab 1985; 61: 990–992. 19. Dunaif A. Insulin resistance and ovarian hyperandrogenism. The Endocrinologist 1992; 2: 248–260. 20. Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis Endoc. Reviews 1997; 18061: 774–800. 21. Dunaif A., Givens J.R., Haseltine F., Merriam G.R. (eds) The Polycystic Ovary Syndrome. Blackwell Scientific, Cambridge. MA. 1992. 22. Dunaif A., Graf M., Mandeli J., Laumas V., Dobrjansky A. Characterization of groups of hyperandrogenic women with acanthosis nigricans, impaired glucose tolerance and or hyperinsulinemia. J Clin Endocrinol Metab 1987; 65: 499–507. 23. Dunaif A., Green G., Phelps R.G., Lebwohl M., Futterweit W., Lewy L. Acanthosis nigricans, insulin action, and hyperandrogenism: clinical, histological, and biochemical findings. J Clin Endocrinol Metab 1991; 73: 590–595. 24. Dunaif A., Hoffman A.R. Insulin resistance and hyperandrogenism: clinical syndromes and possible mechanisms. In: Pancheri P., Zichella L. (eds) Biorhythms and Stress in the Physiopathology of Reproduction. Hemisphere Publishing Co, Washington, DC. 1988; 293–317. 25. Dunaif A., Hoffman A.R., Scully R.E., Flier J.S., Longcope C., Levy L.J., Crowley Jr. W.F. The clinical, biochemical and ovarian morphologic features in women with acanthosis nigricans and masculinization. Obstet Gynecol 1985; 66: 545–552. 26. Dunaif A., Xia J., Book C., Schenker E., Tang Z. Excessive insulin receptor serine phosphorylation in cultured fibroblasts and skeletal muscle: a potential mechanism for insulin resistance in the polycystic ovary syndrome. J Clin Invest 1995; 96: 801–810. 27. Ebina Y., Ellis L., Jarnagin K., Edery M., Graf L., Clauser E., Ou J., Masiarz F., Kan Y.W., Goldfine I.D., Roth R.A., Rutter W.J. The human insulin receptor cDNA: the structural basis for hormoneactivated transmembrane signalling. Cell 1985; 40: 747–758. 28. Ehrmann D.A., Barnes R.B., Rosenfield R.L. Polycystic ovary syndrome as a form of functional ovarian hyperandrogenism due to dysregulation of androgen secretion. Endocr Rev 1995; 16: 322–353. 29. Ehrmann D.A., Rosenfield R.L., Barnes R.B., Brigell D.F., Sheikh Z. Detection of functional ovarian hyperandrogenism in women with androgen excess. N Engl J Med 1992; 327: 157–162. 30. Flier J.S., Eastman R.C., Minaker K.L., Matteson D., Rowe J.W. Acanthosis nigricans in obese women with hyperandrogenism. Characterization of an insulin-resistant state distinct from the Type A and B syndromes. Diabetes 1985; 34: 101–107. 31. Flier J.S., Kahn R., Roth J., Bar R.S. Antibodies that impair insulin receptor binding in an unusual diabetic syndrome with severe insulin resistance. Science 1975; 190: 63–65. 32. Franks S. Polycystic ovary syndrome. N Engl J Med 1995; 333: 853–861. 33. Frattali A.L., Treadway J.L., Pessin J.E. Transmembrane signaling by the human insulin receptor kinase. Relationship between intramolecular beta-subunit trans- and cis- autophosphorylation and substrate kinase activation. J Biol Chem 1992; 267: 19521–19528. 34. Froesch E.R., Zapf J. Insulin-like growth factors and insulin comparative aspects. Diabetologia 1985; 28: 485–493. 35. Futterweit W., Dunaif A., Yeh H., Kingsley P. The prevalence of hyperandrogenism in 109 consecutive women presenting with diffuse alopecia. J Am Acad Dermatol 1988; 19: 831–836. 36. Goldzieher J.W., Green J.A. The polycystic ovary. I. Clinical and histologic features. J Clin Endocrinol Metab 1962; 22: 325–338. 37. Guo H., Damuni Z. Autophosphorylation-activated protein kinase phosphorylates and inactivates protein phosphatase 2A. Proc Natl Acad Sci USA 1993; 90: 2500–2504. 38. Hintz R.L., Thorsson A.V., Enberg G., Hall K. IGF-II binding on human lymphoid cells: demonstration of a common high affinity receptor for insulin like peptides. Biochem Biophys Res Commun 1984; 118: 774–782. 39. Holte J., Bergh T., Berne C., Berglund L., Lithell H. Enhanced early insulin response to glucose in relation to insulin resistance in women with polycystic ovary syndrome and normal glucose tolerance. J Clin Endoc and Metab 1994; 78: 1052–1058. 40. Hotamisligil G.S., Peraldi P., Budavari A., Ellis R., White M.F., Spiegelman B.M. IRS-1 mediated inhibition of insulin receptor tyrosine kinase activity in TNF and obesity-induced insulin resistance. Science 1996; 271: 665–668. 41. Hughesdon P.E. Morphology and morphogenesis of the Stein-Leventhal ovary and of so-called “hyperthecosis”. Obstet Gynecol Sury 1982; 37: 59–77. 42. Hull M.G.R. Epidemiology of infertility and polycystic ovarian disease: endocrinological and demographic studies. Gynecol Endocrinol 1987; 1: 235–245. 43. Jonas H.A., Cox A.J., Harrison L.C. Delineation of atypical insulin receptors from classical insulin and type I insulin-like growth factor receptors in human placenta. Biochem J 1989; 257: 101–107. 44. Judd H.L., Scully R.E., Herbst A.L., Yen S.S.C., Ingersol F.M., Kliman B. Familial hyperthecosis: comparison of endocrinologic and histologic findings with polycystic ovarian disease. Am J Obstet Gynecol 1973; 117: 976–982. 45. Kahn C.R. Insulin action, diabetogenes, and the cause of Type II diabetes. Diabetes 1994; 43: 1066–1084. 46. Kahn C.R. The molecular mechanism of insulin action. Annu Rev Med 1985; 36: 429–451. 47. Kahn C.R., Flier J.S., Bar R.S., Archer J.A., Gorden P., Martin M.M., Roth J. The syndromes of insulin resistance and acanthosis nigricans. N Engl J Med 1976; 294: 739–745. 48. Karasik A., Rothenberg P.L., Yamada K., White M.F., Kahn C.R. Increased protein kinase C activity is linked to reduced insulin receptor autophosphorylation in liver of starved rats. J Biol Chem 1990; 265: 10226–10230. 49. Kasuga M., Hedo J.A., Yamada K.M., Kahn C.R. The structure of the insulin receptor and its subunits: evidence for multiple nonreduced forms and a 210 kD possible proreceptor I. Biol Chem 1982; 257: 10392–10399. 50. Kasuga M., Karlsson F.A., Kahn C.R. Insulin stimulates the phosphorylation of the 95,000 – dalton subunit of its own receptor. Science 1982; 215: 185–187. 51. Keettel W.C., Bradbury J.T., Stoddard F.J. Observations on the PCO syndrome. Am J Obstet Gynecol 1957; 73: 954–65. 52. Kierland R.R., Lakatos I., Szijarto L. Acanthosis nigricans: An analysis of data in twenty-two cases and a study of its frequency in necropsy material. J Invest Dermatol 1947; 9: 299–305. 53. Leroith D., Werner H., Beitner-Johnson D., Roberts Jr. C.T. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev 1995; 16: 143–163. 54. Lobo R.A., Goebelsmann U., Horton R. Evidence for the importance of peripheral tissue events in the development of hirsutism in polycystic ovary syndrome. J Clin Endocrinol Metab 1983; 57: 393–397. 55. McKenna T.J., Moore A., Magee F., Cunningham S. Amenorrhea with cryptic hyperandrogenemia. J Clin Endocrinol Metab 1983; 56: 893–896. 56. Mechanick J., Dunaif A. Masculinization: a clinical approach to the diagnosis and treatment of hyperandrogenic women. In: Mazzaferri E (ed) Advances in Endocrinology and Metabolism. Mosley Year Book, Inc, Chicago, Il. 1990; Pp 129–173. 57. Morales A.J., Laughlin G.A., Butzow T., Maheshwari H., Baumann G., Yen S.S.C. Insulin, somatotropic, and luteinizing hormones axes in lean and obese women with polycystic ovary syndrome: common and distinct features. J Clin Endocrinol Metab 1996; 81: 2854–2864. 58. Moxham C.P., Duronio V., Jacobs S. Insulin-like growth factor I receptor beta-subunit heterogeneity. J Biol Chem 1989; 264: 13238–13244. 59. Moxham C.P., Jacobs S. Insulin / IGF-I receptor hybrids: a mechanism for increasing receptor diversity. J Cell Biochem 1992; 48: 136–140. 60. Nagamani M., Dinh T.V., Kelver M.E. Hyperinsulinemia in hyperthecosis of the ovaries. Am J Obstet Gynecol 1986; 154: 384–389. 61. Nestler J.E., Strauss III J.F. Insulin as an effector of human ovarian and adrenal steroid metabolism. Endocrinol Metab Clin North Am 1991; 20: 807–823. 62. O’Rahilly S., Woong H.C., Patel P., Turner R.C., Flier J.S., Moller D.E. Detection of mutations in insulin-receptor gene in NIDDM patients by analysis of single-stranded conformation polymorphisms. Diabetes 1991; 40: 777–782. 63. Pasquali R., Venturoli S., Paradisi R., Capelli M., Parenti N., Melchionda N. Insulin and C-peptide levels in obese patients with polycystic ovaries. Horm Metab Res 1982; 14: 284–287. 64. Patti M.E., Sun X.J., Bruening J.C., Araki E., Lipes M.A., White M.F., Kahn C.R. 4PS/insulin receptor substrate (IRS-2) is the alternative substrate of the insulin receptor in IRS-1 deficient mice. J Biol Chem 1995; 270: 24670–24673. 65. Peters E.J., Stuart C.A., Prince M.J. Acanthosis nigricans and obesity: acquired and intrinsic defects in insulin action. Metabolism 1986; 35: 807–813. 66. Polson D.W., Wadsworth J., Adams J., Franks S. Polycystic ovaries – a common finding in normal women. Lancet 1988; 1: 870–872. 67. Poretsky L. On the paradox of insulin-induced hyperandrogenism in insulin-resistant statets. Endocr Rev 1991; 12: 3–13. 68. Poretsky L., Kalin M.F. The gonadotropic function of insulin. Endocr Rev 1987; 8: 132–141. 69. Quintana B., Chinchilli V., Sieber J., Fultz P., George N., Dunaif A. High risk of glucose intolerance (Gl) in women with oligomenorrhea or with polycystic ovary syndrome (PCOS). Program of the 77th Annual Meeting of The Endocrine Society, Washington DC, 1995; (Abstract OR3-5: 50). 70. Rebar R., Judd H.L., Yen S.S., Rakoff J., Vandenberg G., Naftolin F. Characterization of the inappropriate gonadotropin secretion in polycystic ovary syndrome. J Clin Invest 1976; 57: 1320–9. 71. el-Roeiy A., Chen X., Roberts V.J., LeRoith D., Roberts Jr. C.T., Yen S.S.C. Expression of insulin-like growth factor-I (IGF-I) and IGF-II and the IGF-I, IGF-II, and insulin receptor genes and localization of the gene products in the human ovary. J Clin Endocrinol Metab 1993; 77: 1411–1418. 72. el-Roeiy A., Chen X., Roberts V.J., Shimasakai S., Ling N., LeRoith D., Roberts Jr. C.T., Yen S.S.C. Expression of the genes encoding the insulin-like growth factors (IGF-I and II), the IGF and insulin receptors, and IGF-binding proteins –1–6 and the localization of their gene products in normal and polycystic ovary syndrome ovaries. J Clin Endocrinol Metab 1994; 78: 1488–1496. 73. Santen R.J., Bardin C.W. Episodic luteinizing hormone secretion in man. Pulse analysis, clinical interpretation, physiologic mechanisms. J Clin Invest 1973; 52: 2617–28. 74. Shoelson S.E., Boni-Schnetzler M., Pilch P.F., Kahn C.R. Autophosphorylation within insulin receptor beta-subunits can occur as an intramolecular process. Biochemistry 1991; 30: 7740–7746. 75. Shoupe D., Kumar D.D., Lobo R.A. Insulin resistance in polycystic ovary syndrome. Am J Obstet Gynecol 1983; 147: 588–592. 76. Stuart C.A., Peters E.J., Prince M.J., Richards G., Cavallo A., Meyer W.J. Insulin resistance with acanthosis nigricans: the roles of obesity and androgen excess. Metabolism 1986; 35: 197–205. 77. Sun X.J., Rothenberg P., Kahn C.R., Backer J.M., Araki E., Wilden P.A., Cahill D.A., Goldstein B.J., White M.F. Structure of the insulin receptor substrate IRS-I defines a unique signal transduction protein. Nature 1991; 352: 73–77. 78. Sun X.J., Wang L.M., Zhang Y., Yenush L., Myers Jr. M.G., Glasheen E., Lane W.S., Pierce J.H., White M.F. Role of IRS-2 in insulin and cytokine signalling. Nature 1995; 377: 173–177. 79. Takayama S., White M.F., Kahn C.R. Phorbol ester-induced serine phosphorylation of the insulin receptor decreases its tyrosine kinase activity. J Biol Chem 1988; 263: 3440–3447. 80. Taylor S.I., Cama A., Accili D., Barbetti F., Quon J., Sierra M.D.L.L., Suzuki Y., Koller E., Levy-Toledano R., Wertheimer E., Moncada V.Y., Kadowaki T. Mutations in the insulin receptor gene. Endocr Rev 1992; 13: 566–595. 81. Theroux S.J., Latour D.A., Stanley K., Raden D.L., Davis R.J. Signal transduction by the epidermal growth factor receptor is attenuated by a COOH-terminal domain serine phosphorylation site. J Biol Chem 1992; 267: 16620–16626. 82. Ullrich A., Bell J.R., Chen E.Y., Herrera R., Petruzzelli L.M., Dull T.J., Gray A., Coussens L., Liao Y-C., Tsubokawa M., Mason A., Seeburg P.H., Grunfeld C., Rosen O.M., Ramachandran J. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 1985; 313: 756–761. 83. Ullrich A., Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990; 61: 203–212. 84. Waldstreicher J., Santoro N.F., Hall J.E., Filicori M., Crowley Jr. W.F. Hyperfunction of the hypothalamic-pituitary axis in women with polycystic ovarian disease: indirect evidence for partial gonadotroph desensitization. J Clin Endocrinol Metab 1988; 66: 165–172. 85. Willis D., Franks S. Insulin action in human granulosa cells from normal and polycystic ovaries is mediated by the insulin receptor and not the type-I insulin-like growth factor receptor. J Clin Endocrinol Metab 1995; 80: 3788–3790. 86. Willis D., Mason H., Gilling-Smith C., Franks S. Modulation by insulin of follicle-stimulating hormone and luteinizing hormone actions in human granulosa cells of normal and polycystic ovaries. J Clin Endocrinol Metab 1996; 81: 302–309. 87. Yen S.S.C. The polycystic ovary syndrome. Clin Endocrinol (Oxf) 1980; 12: 177–208. 88. Yen S.S.C., Vela P., Rankin J. Inappropriate secretion of follicle-stimulating hormone and luteinizing hormone in polycystic ovarian disease. J Clin Endocrinol Metab 1970; 30: 435–442 89. Myers Jr. M.G., Sun X.J., White M.F. The IRS-I signaling system. Trends Biochem Sci 1994; 19: 289–293. 90. Dunaif A. Polycystic ovary syndrome and obesity. In: Byorntorp P., Brodoff B.N. (eds) Obesity. J.B. Lippincott Co., Philadelphia, 1992; 594–605. |