Синдром тестикулярной феминизации: клиническое, гормональное и молекулярно-генетическое исследование.
|
Синдром тестикулярной феминизации (androgen insensitivity syndrome) представляет собой полную или частичную нечувствительность тканей к андрогенам, обусловленную нарушением связывающей способности рецептора андрогенов или пострецепторным дефектом. Синдром наследуется по Х-сцепленному рецессивному типу [1, 2, 8, 10, 15].
СТФ характеризуется кариотипом 46,XY, гонадами и внутренними гениталиями мужского типа, широкой вариабельностью строения наружных половых органов. Выделяют полную и неполную формы синдрома [8]. При полной форме синдрома наружные гениталии имеют правильное женское строение, при неполной форме - от интерсексуального до правильного мужского [1, 2, 10, 15]. Гормональный статус больных с СТФ характеризуется высоким уровнем гонадотропинов, с преимущественным повышением уровня лютеинизирующего гормона (ЛГ), высоким уровнем тестостерона (Т), и умеренно повышенным уровнем эстрадиола (Е2). Указанные гормональные особенности наблюдаются преимущественно у больных с полной формой СТФ. При неполной форме синдрома изменения гормонального статуса выражены в меньшей степени [2, 10, 15]. До внедрения в клиническую практику методов молекулярно-генетического анализа, диагностика СТФ основывалась на результатах клинико-генеалогического, цитогенетического и гормонального методов обследования. Для подтверждения диагноза был разработан метод определения связывающей способности рецепторов андрогенов в фибробластах кожи гениталий, однако диагностическая ценность этого метода невысока [15]. В восьмидесятых годах была установлена локализация гена, ответственного за развитие болезни, на хромосоме Х в Хq11-13 [12]. Ген получил название AR (androgen receptor). Он кодирует рецепторный белок, необходимый для воздействия андрогенов на ткани. Ген экспрессируется в клетках многих органов человека на протяжении всего онтогенеза, поддерживая количество рецепторного белка на необходимом уровне. В эмбриогенезе AR регулирует правильное развитие вольфовых протоков и формирование наружных половых органов по мужскому типу; в пубертатном периоде - развитие вторичных половых признаков и созревание сперматогенного эпителия [8, 15]. Таким образом, белок AR опосредует действие андрогенов на внутриклеточные механизмы, ответственные за развитие мужских половых признаков и фертильность. Рецепторный белок, кодируемый геном AR, имеет три функционально значимые области: амино-концевую, ДНК-связывающую и взаимодействующую с гормоном (стероидсвязывающую). По данным ряда авторов [8, 10, 15], у больных с СТФ мутации выявляются во всех областях гена AR с преимущественной локализацией в ДНК-связывающей и стероидсвязывающей. Спектр мутаций гена AR при СТФ достаточно разнообразен, однако наиболее частыми являются миссенс-мутации, доля которых составляет 80%. Молекулярно-генетический анализ гена AR имеет важное практическое значение. Идентификация мутаций гена при наличии характерной клинической картины подтверждает диагноз СТФ с вероятностью, близкой к 100%. Кроме того, исследование гена AR может быть использовано для проведения пренатальной диагностики и выявления гетерозиготного носительства в семьях, отягощенных СТФ [11, 15]. Материал и методы В клинике проведено комплексное обследование семьи, имеющей двух сибсов с неполной формой СТФ. Первоначально диагноз был установлен на основании интерсексуального строения наружных гениталий, гормонального исследования, данных цитогенетического анализа, а затем подтвержден молекулярно-генетическим исследованием ДНК больных. У обследуемых выясняли особенности семейного анамнеза, оценивали параметры физического развития, характер и сроки появления вторичных половых признаков, особенности строения наружных гениталий, месторасположение и размеры гонад. Рост больных оценивали с помощью перцентильных таблиц [16], стадию полового развития - согласно классификации Tanner [16], костный возраст - по методу Greulich и Pyle [9]. Базальные уровни гонадотропинов: ЛГ и ФСГ определяли радиоиммунофлюоресцентным методом с использованием стандартных наборов "Иммунотекс" (Чехия). Уровни Е2, Т, дегидроэпиандростерона-сульфата (ДГЭА) исследовали электрохемилюминесцентным методом на аппарате "Элексис", стандартными наборами "Элексис" (ФРГ-Япония). Гормональные исследования проводили в лаборатории "НЦ ЭФиС". Цитогенетическое исследование проводили на культуре лимфоцитов периферической крови. Ультрасонографию органов малого таза, яичек, простаты проводили трансабдоминальным поперечным и продольным сканированием с использованием секторальных и конвексных датчиков с частотой 3 и 5 мГц сонографами "Sim 5000" (Италия) и "Aloka 630" (Япония) в режиме реального времени. Исследование проведено профессором РМАПО М.И. Пыковым. Молекулярная диагностика структурных дефектов гена AR проводилась в лаборатории ДНК-диагностики МГНЦ РАМН. ДНК матери и двух сибсов была выделена из клеток венозной крови путем фенол-хлороформной экстракции с последующим осаждением этанолом. Методом полимеразной цепной реакции (ПЦР) были синтезированы фрагменты ДНК, соответствующие семи экзонам гена AR с прилегающими интронными областями. Полученные фрагменты подвергнуты SSCP (Single Strand Conformation Polymorfism) анализу [14]. Диагностическая ценность SSCP заключается в отборе фрагментов, имеющих точечную мутацию, которая изменяет их электрофоретическую подвижность. Фрагменты с измененной электрофоретической подвижностью секвенированы для установления характера мутации. Результаты Сибс I Ребенок от первой нормально протекавшей беременности, первых срочных родов. Масса тела при рождении 3900 г, длина тела 52 см. При рождении отмечалось неправильное строение наружных половых органов: двусторонний крипторхизм, расщепление мошонки, промежностная гипоспадия, искривление и недоразвитие кавернозных тел, имеющих длину около 1 см. Зарегистрирован и воспитывался в мужском поле. В возрасте 3 лет проведено цитогенетическое исследование. Кариотип - 46,ХY. С этого возраста и в течение последующих 11 лет ребенку проведен ряд последовательных этапов маскулинизирующей пластики наружных гениталий. В возрасте 12 лет появились признаки спонтанного пубертата: половое оволосение, увеличение полового члена. Наряду с этим наблюдалось развитие двусторонней гинекомастии. В 13 лет, в связи с резким увеличением грудных желез до Ма3, ребенку была проведена двусторонняя мастэктомия. Данные обследования в 17 лет: рост 179 см, вес 65 кг, SDS роста +1,8, костный возраст соответствует 17 годам, телосложение мужского типа. Половой статус: Аґ2 Р3, длина полового члена 7 см, наружное отверстие уретры открывается на стволе полового члена. Яички дряблые, располагаются в мошонке, объем правого яичка 5 мл, левого - 4 мл. Эрекции спонтанные и ситуационно обусловленные (1-2 раза в неделю). Таким образом, пубертат начался в нормальные сроки, однако активность процесса полового развития была недостаточна и на момент обследования половое развитие отставало от средневозрастной нормы. Уровень гонадотропинов соответствовал возрастной норме: ЛГ - 9,5 мЕ/л (норма 0,5-12 мЕ/л), ФСГ - 3,8 мЕ/л (норма 0,8-13 мЕ/л). Уровни Т и Е 2 несколько повышены: Т - 36 нмоль/л (норма 7,0-20,0 нмоль/л), Е2 - 0,2 нмоль/л (норма 0,011-0,15 нмоль/л). Уровень ДГЭА соответствовал нормальным показателям - 1,8 мкг/мл (норма: 0,6-2,5 мкг/мл). Данные УЗИ органов малого таза и гонад: яички - в мошонке. Объем правого яичка 2,3 мл, придаток не определяется, паренхима средней эхогенности, однородная. Объем левого яичка 2,0 мл, головка придатка не определяется. Предстательная железа гипоплазирована, паренхима не изменена, семенные пузырьки не определяются. Сибс II Ребенок от второй беременности, вторых срочных родов. Масса тела при рождении 4100 г, длина тела 55 см. С рождения отмечалось неправильное строение наружных половых органов: расщепление мошонки, двусторонний крипторхизм в форме паховой ретенции, в сочетании с водянкой оболочек яичка справа, пенис с удовлетворительно развитыми кавернозными телами, длиной 1 см, промежностная форма гипоспадии. Зарегистрирован и воспитывался в мужском поле. В возрасте 2 мес проведен цитогенетический анализ. Кариотип - 46,ХY. В возрасте от 2 до 13 лет проведен ряд последовательных этапов пластики наружных гениталий. В возрасте 12,5 лет отмечалось появление признаков спонтанного пубертата: половое оволосение, увеличение полового члена, а также двусторонняя гинекомастия. Данные обследования в 16 лет: рост 176 см, вес 60 кг, SDS роста +1,7, костный возраст соответствует 16 годам. Телосложение астеническое со слабо выраженными чертами маскулинизации, половой статус: Ма2 Аґ2 Р4, длина полового члена 6 см. Яички в мошонке, дряблой консистенции, объем правого яичка 4 мл, левого - 4 мл. Наружное отверстие уретры открывается на стволе верхней трети полового члена, в средней трети пениса открывается точечный свищ, эрекции - спонтанные. Таким образом, у сибса 2, как и у сибса 1, пубертат манифестировал в нормальные сроки, однако в дальнейшем адекватного прогресса полового развития не отмечалось. При исследовании гормонального профиля в 16 лет уровень гонадотропинов соответствует возрастной норме: ЛГ - 7,9 мЕ/л (норма 0,5-12 мЕ/л), ФСГ - 2,9 мЕ/л (норма 0,8-13 мЕ/л). Уровень Т повышен - 28,9 нмоль/л (норма 7,0-20,0 нмоль/л). Уровни ДГЭА и Е 2 соответствуют половозрастным нормативам: ДГЭА - 2,2 мкг/мл (норма 0,6-2,5 мкг/мл), Е2 - 0,09 нмоль/л (норма 0,011-0,15 нмоль/л). Данные ультразвукового исследования органов малого таза и гонад: левое яичко - 1,97 мл, паренхима средней эхогенности, однородная, головка придатка определяется плохо. Правое яичко - 2,0 мл, паренхима средней эхогенности, однородная, придаток не определяется. Гемодинамика с обеих сторон не нарушена. Предстательная железа гипоплазирована, без структурных изменений, капсула тонкая. Данные молекулярно-генетического обследования: у обоих больных обнаружено изменение электофоретической подвижности фрагментов второго экзона гена AR (см. рисунок). При секвенировании идентифицирована мутация, представляющая собой замену гуанина (G) на аденин (А) в 2064 положении нуклеотидной последовательности [12]. Гуанин является первым нуклеотидом триплета GGG, кодирующего аминокислоту глицин (Gly (G)), расположенную в 568 положении аминокислотной цепи белка AR. Замена гуанина на аденин приводит к появлению триплета АGG, в результате чего при трансляции вместо глицина встраивается аминокислота аргинин (Arg (R)). Данная мутация является миссенс-мутацией и, в соответствии со стандартной системой записи, обозначается как G568R [4]. Аналогичное изменение гена AR было выявлено при исследовании ДНК матери сибсов. Обсуждение Различные формы ложного мужского гермафродитизма имеют сходную клиническую картину [1, 10]. Дифференциальная диагностика между ними достаточно сложна, особенно в допубертатный период. С целью установления точного диагноза используются генеалогический анамнез, особенности гормонального и клиническо го статуса, особенности течения пубертата [1, 2, 10].
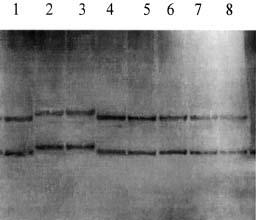
Рисунок. Результат SSCP-анализа 2-го экзона гена AR больных с синдромом тестикулярной феминизации. Дорожки 1, 4-7, 8 - образцы одноцепочечных фрагментов ДНК с неизмененной электрофоретической подвижностью. Дорожки 2 и 3 - образцы одноцепочечных фрагментов ДНК сибсов 1 и 2 соответственно. Электрофоретическая подвижность изменена в обоих образцах. У наших пациентов интерсексуальное строение наружных гениталий, гинекомастия, гипоплазированная предстательная железа, небольшой объем тестикул, кариотип 46,XY, а также высокий уровень тестостерона и эстрадиола, позволили предположить наличие неполной формы СТФ. Подтверждение диагноза было получено после проведения молекулярно-генетического анализа гена AR. У больных с СТФ описано более 240 различных мутаций гена AR [8, 10, 15]. Однако мутаций, которые встречаются с высокой частотой, не выявлено. В связи с этим для каждого больного с СТФ или семьи с несколькими пораженными проводится поиск "индивидуальной" мутации. Самой частой причиной развития СТФ являются различные миссенс-мутации. Их доля среди всех мутаций гена AR составляет 80% [15]. Миссенс-мутации выявлены в разных участках гена, но преимущественно локализуются в стероид-связывающей области. Достоверной зависимости между локализацией миссенс-мутации и фенотипическим проявлением болезни еще не установлено в связи с малым количеством наблюдений для каждой из них [8, 15]. Обнаруженная нами мутация также является миссенс-мутацией. Ранее в триплете 568 были описаны две миссенс-мутации: G568V (замена глицина на валин) и G568W (замена глицина на триптофан) [5, 11]. Обе мутации выявлены у больных с неполной формой СТФ. Обнаруженная нами мутация пополняет число мутаций 568-го кодона. Сходство клинических проявлений у всех больных с мутацией в этом кодоне свидетельствует о частичной сохранности функциональной активности рецепторного белка. Триплет 568 расположен во втором экзоне, который кодирует часть ДНК-связывающей области рецепторного белка AR. ДНК-связывающая область рецептора имеет вторичную конфигурацию, получившую название "цинковые пальцы" [7, 15]. Каждый "цинковый палец" образуется за счет связывания четырех цистеинов, расположенных на небольшом расстоянии друг от друга ионом цинка. Такая пространственная конфигурация характерна для белков, взаимодействующих с регуляторными участками молекулы ДНК. В связи с этим рецепторный белок AR рассматривают как регулятор транскрипции генов половой дифференцировки. Миссенс-мутации ДНК-связывающей области не влияют на связь рецептора с андрогеном, но, по-видимому, снижают его транскрипционную активность, что приводит к развитию СТФ без изменения связывающей способности рецептора андрогенов [5]. Таким образом, можно предположить, что нарушение маскулинизации наружных гениталий и развития половых признаков в период пубертата у сибсов в данной семье обусловлено частичной потерей чувствительности тканей к андрогенам, за счет нарушения транскрипции генов, участвующих в дифференцировке пола по мужскому типу. По-видимому, для функциональной активности белка AR данная мутация не является критической и не приводит к полной потери функции рецептора. Сходство клиники у обследованных больных объясняется идентичностью мутационных изменений гена AR. Неполную форму СТФ необходимо дифференцировать с другими формами ложного мужского гермафродитизма [10]. Большинство из них имеют аутосомно-рецессивный тип наследования, в то время как СТФ наследуется по Х-сцепленному рецессивному типу. Поэтому установление Х-сцепленного рецессивного типа наследования болезни в семье является одним из важнейших критериев диагностики неполной формы СТФ. Однако это возможно далеко не во всех случаях. Так, в семьях с одним пораженным или несколькими пораженными в одном поколении можно предполагать аутосомно-рецессивный тип наследования, а в семьях с одним пораженным также мутацию de novo, которая встречается в 1/3 случаев СТФ. В связи с этим ДНК-диагностика позволяет подтвердить диагноз СТФ у больного и выявить гетерозиготных носителей мутации в семье. В нашем случае идентификация мутации G568R в гене AR у больных позволила подтвердить диагноз СТФ. Обследование матери сибсов показало, что данную мутацию они унаследовали от нее. Результаты молекулярно-генетического исследования могут быть использованы для пренатальной диагностики. Работа выполнена в рамках подпрограммы «Национальные приоритеты в медицине и здравоохранении» программы «Геном человека». Г.Р. Осипова, Э.П. Касаткина, А.Ю. Дергачева, Ю.В. Лозовая, Я.В. Кононова, А.В. Поляков Российская медицинская академия последипломного образования Минздрава РФ, Медико-генетический научный центр РАМН, Москва Литература 1. Касаткина Э.П. Реабилитация больных гермафродитиз мом. Материалы республиканского совещания-семинара главных детских эндокринологов, 12-13 октября, Смоленск 1999; 132-144. 2. Самсонова Л.Н. Оптимизация дифференциальной диагностики вариантов гермафродитизма: Автореф. дис. ... канд. мед. наук. М 1996. 3. Batch J.A., Williams D.M., Davies H.R. et al. Role of the androgen receptor in male sexual differentiation. Horm Res 1992; 38: 226-229. 4. Beudet A.L., Tsui L.-C. A suggested nomenclature for designing mutations. Human Mutat 1993; 3: 1662-1666. 5. Chang Y.T., Migeon C.J., Brown T.R. Human androgen insensitivity due to androgen receptor gene point mutation in subjects with normal androgen receptor levels but impaired biological activity. Program of the 73rd Annual Meeting of the Endocrine Society, Washington D.C. 1991; 37. 6. De Bellis A., Quigley C.A., Wilson E.M. Single base mutations in the androgen receptor gene cousin complete androgen insensitivity: rapid detection by a modified denaturing gradient gel electrophoresis technique. J Mol Endocrinol 1992; 6: 1909-1920. 7. Freedman L.P. Anatomy of the steroid receptor zinc finger region. Endocrinol Rev 1992; 13: 129-145. 8. Gottleb B., Pinsky L., Lenore K.B. Androgen Insensitivity. Am J Med Genet 1999; 89: 210-217. 9. Greulich W.W., Pyle S.I. Radiographic atlas of skeletal development of the hand and wrist. 2 nd Ed. Stanford: Stanford University press 1959. 10. Wilson J.D., Foster D.W., Kronenberg H.M., Larsen P.R. Williams textbook of endocrinology. W.B. Sounders company 1998; 1331-1405. 11. Lobaccaro J.M., Belon C., Lubroso S. et al. Molecular prenatal diagnosis of partial androgen insensitivity syndrome based on the Hind III polymorphism of the androgen receptor gene. Clin Endocrinol 1994; 40: 297-302. 12. Lubahn D.B., Joseph D.R., Sullivan P.M. et al. Cloning human androgen receptor: complementary DNA and localization to the X chromosome. Science 1989; 240: 327-330. 13. McPhaul M.J., Marcelli M., Zoppi S. et al. Mutations in the ligand-binding domain of the androgen receptor gene cluster in two regions of the gene. J Clin Invest 1992; 90: 2097-2101. 14. Orita M., Iwahana H., Kanazawa H., Hayashi K., Sekiya T. Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. PNAS USA 1989; 86: 2766-2770. 15. Quigley C.A., Bellis A., Marschke K.B. et al. Androgen receptor defects: Historical, clinical, and molecular perspectives. Endocrine reviews 1995; 16: 271-321. 16. Tanner J.M., Whitehouse R. Clinical longitudinal standards for height, weight, height velocity, and the stages of puberty. Arch Dis Child 1976; 51: 170-179. 17. Weidmann W., Peters B., Romalo G. Response to androgen treatment in patients with Partial Androgen Insensitivity and a mutation in Deoxyribonucleic Acid-Binding Domain of Androgen Receptor. J Clin Endocrinol Metab 1998; 83: 1173-1178. |